2024-11-01 ノースウェスタン大学
<関連情報>
- https://news.northwestern.edu/stories/2024/11/new-huntingtons-treatment-prevents-protein-aggregation/
- https://www.science.org/doi/10.1126/sciadv.ado8307
- https://www.nature.com/articles/ncomms12646
タンパク質模倣ポリマーがミトコンドリア障害をブロックし、ハンチントン病ニューロンを救う Proteomimetic polymer blocks mitochondrial damage, rescues Huntington’s neurons, and slows onset of neuropathology in vivo
Wonmin Choi, Mara Fattah, Yutong Shang, Matthew P. Thompson, […], and Nathan C. Giannesch
Science Advances Published:1 Nov 2024
DOI:https://doi.org/10.1126/sciadv.ado8307
")
Abstract
Recently, it has been shown that blocking the binding of valosin-containing protein (VCP) to mutant huntingtin (mtHtt) can prevent neuronal mitochondrial autophagy in Huntington’s disease (HD) models. Herein, we describe the development and efficacy of a protein-like polymer (PLP) for inhibiting this interaction in cellular and in vivo models of HD. PLPs exhibit bioactivity in HD mouse striatal cells by successfully inhibiting mitochondrial destruction. PLP is notably resilient to in vitro enzyme, serum, and liver microsome stability assays, which render analogous control oligopeptides ineffective. PLP demonstrates a 2000-fold increase in circulation half-life compared to peptides, exhibiting an elimination half-life of 152 hours. In vivo efficacy studies in HD transgenic mice (R6/2) confirm the superior bioactivity of PLP compared to free peptide through behavioral and neuropathological analyses. PLP functions by preventing pathologic VCP/mtHtt binding in HD animal models; exhibits enhanced efficacy over the parent, free peptide; and implicates the PLP as a platform with potential for translational central nervous system therapeutics.
ミトコンドリアへのVCPの動員は、ハンチントン病モデルにおいてマイトファジーの障害と神経変性を引き起こす VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease
Xing Guo,XiaoYan Sun,Di Hu,Ya-Juan Wang,Hisashi Fujioka,Rajan Vyas,Sudha Chakrapani,Amit Umesh Joshi,Yu Luo,Daria Mochly-Rosen & Xin Qi
Nature Communications Published:26 August 2016
DOI:https://doi.org/10.1038/ncomms12646
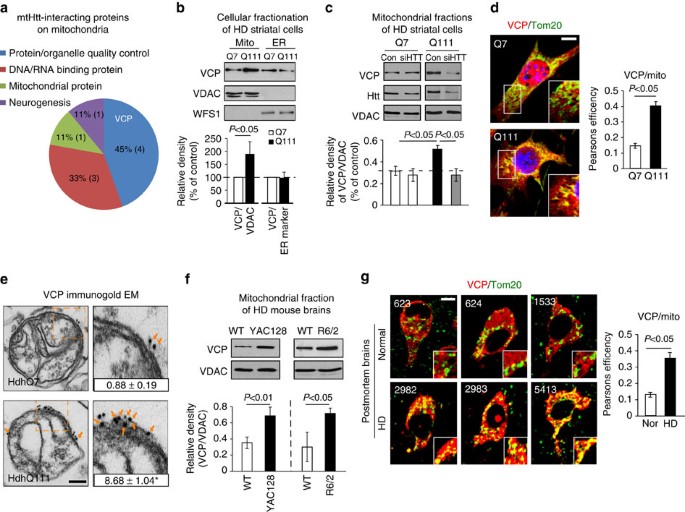
Abstract
Mutant Huntingtin (mtHtt) causes neurodegeneration in Huntington’s disease (HD) by evoking defects in the mitochondria, but the underlying mechanisms remains elusive. Our proteomic analysis identifies valosin-containing protein (VCP) as an mtHtt-binding protein on the mitochondria. Here we show that VCP is selectively translocated to the mitochondria, where it is bound to mtHtt in various HD models. Mitochondria-accumulated VCP elicits excessive mitophagy, causing neuronal cell death. Blocking mtHtt/VCP mitochondrial interaction with a peptide, HV-3, abolishes VCP translocation to the mitochondria, corrects excessive mitophagy and reduces cell death in HD mouse- and patient-derived cells and HD transgenic mouse brains. Treatment with HV-3 reduces behavioural and neuropathological phenotypes of HD in both fragment- and full-length mtHtt transgenic mice. Our findings demonstrate a causal role of mtHtt-induced VCP mitochondrial accumulation in HD pathogenesis and suggest that the peptide HV-3 might be a useful tool for developing new therapeutics to treat HD.
")
")