
2023-06-21 理化学研究所
理化学研究所(理研)計算科学研究センター(R-CCS)HPC/AI駆動型医薬プラットフォーム部門 バイオメディカル計算知能ユニットの德久 淳師 上級研究員、奥野 恭史 ユニットリーダー(HPC/AI駆動型医薬プラットフォーム部門部門長)らの共同研究チームは、タンパク質[1]立体構造推論ソフトウエア「OpenFold」をスーパーコンピュータ「富岳」[2]へ実装し、高速化手法の開発により、超並列環境における高スループット[3]性の達成に成功しました。「富岳」実装版「OpenFold」は、2023年6月21日より、理研R-CSS(https://github.com/RIKEN-RCCS/OpenFold-for-Fugaku)よりオープンソースソフトウエアの形で創薬研究者などへ広く提供され、今後、大量のアミノ酸配列からタンパク質立体構造を高速に推論することが可能になります。
本研究成果は、構造未知のタンパク質立体構造の大規模高速推論を可能とし、分子レベルの知見を通してゲノム医療や次世代創薬への貢献が期待できます。
今回、共同研究チームは、近年英国で開発された、アミノ酸配列からタンパク質の立体構造を高精度に推論するAlphaFold2(AF2)[4]のオープンソースソフトウエアである「OpenFold」を「富岳」へ実装し、超並列環境におけるスループット性を分析しました。今回提供されるソフトウエアには、分析結果に基づいて富士通株式会社が新たに開発した大量のタンパク質を超並列環境で予測するための高速化技術を導入しており、推論全体で技術導入前に比べて6.3倍の高スループット性を実現しています。
本研究は、米国時間6月20日に開催された国際ワークショップ『FlexScience ’23』で発表されました。

「富岳」に実装されたタンパク質立体構造推論ソフトウエア「OpenFold」の概要
背景
生命活動の担い手であるタンパク質は20種類のアミノ酸からなる配列に応じた固有の立体構造を有しており、さまざまな分子機能と密接に関わっています。タンパク質立体構造は創薬において重要な知見を与えるため、これまでに約20万個のタンパク質立体構造が決定されデータベースに登録されてきました。しかし、結晶化困難などの理由により、実験的に構造を決めることが難しく、性質や構造が未知のタンパク質も数多く存在しています。
近年、英国のDeepMind社により開発されたAF2は、実験的に決定された約20万個のタンパク質立体構造とそれに対応するアミノ酸配列を学習することで、構造未知のアミノ酸配列から立体構造モデルを推論します(図1)。
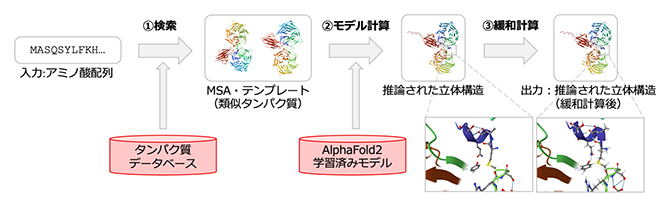
図1 AlphaFold2(AF2)がアミノ酸配列から立体構造を推論する手順
AF2における推論は、アミノ酸配列を入力とし、①タンパク質データベースを用いたMSA[5]・テンプレート検索[6]、②AF2学習済みモデルを用いたモデル計算[7]、③緩和計算[8](物理化学的な構造ゆがみを解消するための分子力場を用いた構造最適化)の手順からなる。
例えば、私たちの腸内には約1,000種の腸内細菌が生息しており、多様な腸内生成物の相互作用が健康維持と密接な関わりを持ちます。その分子メカニズムには、数十万を超える構造未知を含むタンパク質が関与していると考えられています。このような多数のタンパク質の立体構造をAF2により高速に推論できれば、腸内環境を維持する分子メカニズムの解明が進み、腸内環境の改善に有効な物質が同定されることも期待されます。
一方、従来のAF2高速化の研究では、大規模な推論を高速に実現する高スループット性の手法はあまり検討されてきませんでした。AF2は通常、画像処理装置(GPU)などの加速装置を利用して計算速度を向上させますが、本研究では、「富岳」に代表される中央演算処理装置(CPU)超並列環境、つまり大規模な計算資源を活用した高スループット性の達成を目指しました。
研究手法と成果
共同研究チームは、「OpenFold」を「富岳」へ実装し高速化することで、CPU超並列環境におけるスループット性を改善しました。検索タスクにおいて大量のタンパク質の一斉処理技術を開発し、1万件(1万個のタンパク質)の入力アミノ酸配列で評価した結果、高速化する前に比べて平均8.5倍の高速化(スループット増加)を実現しました(図2)。
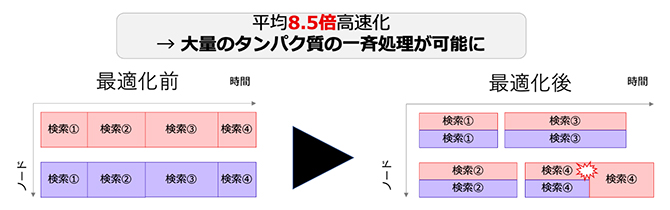
図2 検索タスクにおける高速化手法の概要
検索タスクの実行順序および同時実行数を最適化し、メモリ不足時には計算資源量を自動で再割り当てする機構を構築した。
さらにモデル計算タスクにおいては、性能低下の回避や一部のGPU向け実装をCPU向けに最適化した結果、最適化前に比べて平均1.3倍の高速化を実現し、各タンパク質の高速処理が可能になりました(図3)。

図3 モデル計算タスクにおける高速化手法の概要
バッチ行列積[9]の入力全体をあらかじめ並べ替え、性能低下を回避する工夫をした。加えて、アテンション機構[10]など一部のGPU向け実装をCPUに移植した。
以上のようにして高速化を施した「富岳」実装版「OpenFold」を用いて、1万件の入力アミノ酸配列を評価したところ、推論手順全体で最適化前と比較し6.3倍の高スループット性を達成しました。これは、例えば「富岳」の全系を用いた場合、1時間当たり約120万件のアミノ酸配列を処理する見積もりであり、「富岳」がタンパク質立体構造の大規模高速推論に対して実用的なCPUシステムであることが示されました。
今後の期待
理研R-CCSのHPC/AI駆動型医薬プラットフォーム部門においては、「富岳」への創薬DXプラットフォーム[11]の構築を進めており、今回開発した「富岳」実装版「OpenFold」は、創薬DXプラットフォームにおいて、標的タンパク質の構造を高速に推論する重要な要素技術となると期待できます。
理研は現在、「富岳」のソフトウエアやアプリケーションの成果をArmアーキテクチャ[12]ベースの商業クラウド上でも直接利用できるバーチャル「富岳」の環境整備を進めていますが、今後「富岳」実装版「OpenFold」も同様にクラウド環境で利用可能にし、タンパク質の構造多様性(立体構造の揺らぎ)を考慮した推論ができるように改良を進める予定です。
本成果は、今後、医療や創薬分野のDX化において幅広く活用されるものと期待できます。
補足説明
1.タンパク質
生命活動をつかさどる柔らかな機能性分子。20種類のアミノ酸が複数連なったポリペプチド鎖で、アミノ酸配列に基づき固有の立体構造を取る。機能を発現する際には、構造が変化することが多い。
2.スーパーコンピュータ「富岳」
スーパーコンピュータ「京」の後継機。2020年代に社会的・科学的課題を解決し日本の成長に貢献すること、世界をリードする成果を生み出すことを目的とし、2021年3月に共用が開始された。電力性能、計算性能、ユーザーの利便性・使い勝手の良さ、画期的な成果の創出、ビッグデータやAIの加速機能の総合力において世界最高レベルを誇る。現在、「富岳」は日本が目指すSociety 5.0の実現に不可欠なハイパフォーマンスコンピューティング(HPC)インフラとして活用されている。
3.スループット
コンピュータが単位時間あたりに処理できるデータ量。本研究の場合、処理できるアミノ酸配列数に注目している。
4.AlphaFold2(AF2)
アミノ酸配列からタンパク質の立体構造を予測するAIプログラム。英国にあるGoogle DeepMind社で開発され、2020年に開催されたタンパク質構造予測精密評価 (CASP)コンテストにおいて優勝し、並外れた予測性能を示した。タンパク質の折り畳み問題における驚異的な進歩といえる。
5.MSA
複数のアミノ酸配列に対してギャップを挿入して対応する部分が並ぶように整列し、アミノ酸配列の一致度・類似度を調べる方法。類似性が高いアミノ酸配列を持つタンパク質の機能や構造は似ていることが多いことから、未知のタンパク質の機能や構造に対して示唆を与える。MSAはMultiple Sequence Alignmentの略。
6.テンプレート検索
アミノ酸から立体構造を予測する際に、鋳型となる構造を既知構造のデータベースから検索すること。鋳型構造の使用は任意であり、推論の際に必ずしも必要ではない。
7.モデル計算
全てのアミノ酸残基が原点に集中している構造を初期構造として、アミノ酸配列の特徴を共進化の観点から表すMSA表現、およびアミノ酸残基対の距離の特徴を表すペア表現を手掛かりに、各アミノ残基に対して相対的移動(並進・回転)を繰り返すことで立体構造を得る。
8.緩和計算
物理化学的な構造ゆがみを解消するため、推論した立体構造に対して軽微な分子シミュレーションを実施すること。
9.バッチ行列積
一定量のデータ(バッチ)に対して、まとめて行列の積を計算すること。
10.アテンション機構
ニューラルネットワークの学習法の一つで、認知的な注意を模倣し、データの重要な部分に注意を払う仕組み。
11.創薬DXプラットフォーム
「富岳」上で、創薬プロセスの革新に必要なさまざまなAI・シミュレーションツールをシームレスに活用可能にしたHPC基盤。
12.Armアーキテクチャ
世界で広く使用されているコンピュータの基本設計で、消費電力を抑える特長を持つ。
共同研究チーム
理化学研究所 計算科学研究センター HPC/AI駆動型医薬プラットフォーム部門
バイオメディカル計算知能ユニット
ユニットリーダー 奥野 恭史(オクノ・ヤスシ)
(HPC/AI駆動型医薬プラットフォーム部門 部門長、京都大学大学院 医学研究科 教授)
上級研究員 德久 淳師(トクヒサ・アツシ)
富士通株式会社 コンピューティング研究所
研究員 大山 洋介(オオヤマ・ヨウスケ)
研究員 田渕 晶大(タブチ・アキヒロ)
研究支援
本研究は、理研-富士通共同研究費「HPCとAIを活用した創薬イノベーション」による支援ならびに文部科学省「富岳」高度化・利用拡大枠利用課題「バイオメディカル応用のためのシミュレーション・AI 技術開発による「富岳」の高度化」(課題番号:ra00018)、一般課題「Cryo-EMテンプレートマッチング法による新型コロナウイルス-変異型スパイクタンパク質の構造多形に関する研究」(課題番号:hp220078)、Society 5.0推進利用課題『「富岳」を機軸とした創薬DXプラットフォームの構築』(課題番号:hp220284)の計算資源の提供を受けて実施しました。
原論文情報
Yosuke Oyama, Akihiro Tabuchi, Atsushi Tokuhisa, “Accelerating AlphaFold2 Inference of Protein Three-Dimensional Structure on the Supercomputer Fugaku”, FlexScience ’23, 10.1145/3589013.3596674
発表者
理化学研究所
計算科学研究センター HPC/AI駆動型医薬プラットフォーム部門 バイオメディカル計算知能ユニット
ユニットリーダー 奥野 恭史(オクノ・ヤスシ)
(HPC/AI駆動型医薬プラットフォーム部門 部門長)
上級研究員 德久 淳師(トクヒサ・アツシ)
報道担当
理化学研究所 広報室 報道担当
")
