
2024-08-28 カリフォルニア大学サンディエゴ校(UCSD)
虚血性心疾患は世界的な死因の主要な要因であり、心筋梗塞(MI)によって引き起こされる。MI後の炎症が心不全につながるが、抗炎症薬は効果が薄い。カリフォルニア大学サンディエゴ校の研究者は、「ボーダーゾーン」と呼ばれる心臓周囲の領域で、心筋細胞が核膜破裂を引き起こし、核DNAが漏出し、炎症反応が始まることを発見した。これにより心臓壁が弱まり、心不全の原因となる。この発見は新たな治療法の可能性を示している。
- https://today.ucsd.edu/story/borderzone-breakthrough-a-new-source-of-cardiac-inflammation
- https://www.nature.com/articles/s41586-024-07806-1
- https://www.nature.com/articles/s44161-022-00160-3
- https://www.nature.com/articles/nm.4428
傷害境界域におけるI型インターフェロン反応の空間的集積 Spatially clustered type I interferon responses at injury borderzones
V. K. Ninh,D. M. Calcagno,J. D. Yu,B. Zhang,N. Taghdiri,R. Sehgal,J. M. Mesfin,C. J. Chen,K. Kalhor,A. Toomu,J. M. Duran,E. Adler,J. Hu,K. Zhang,K. L. Christman,Z. Fu,B. Bintu & K. R. King
Nature Published28 August 2024
DOIhttps://doi.org/10.1038/s41586-024-07806-1
")
Abstract
Sterile inflammation after myocardial infarction is classically credited to myeloid cells interacting with dead cell debris in the infarct zone1,2. Here we show that cardiomyocytes are the dominant initiators of a previously undescribed type I interferon response in the infarct borderzone. Using spatial transcriptomics analysis in mice and humans, we find that myocardial infarction induces colonies of interferon-induced cells (IFNICs) expressing interferon-stimulated genes decorating the borderzone, where cardiomyocytes experience mechanical stress, nuclear rupture and escape of chromosomal DNA. Cardiomyocyte-selective deletion of Irf3 abrogated IFNIC colonies, whereas mice lacking Irf3 in fibroblasts, macrophages, neutrophils or endothelial cells, Ccr2-deficient mice or plasmacytoid-dendritic-cell-depleted mice did not. Interferons blunted the protective matricellular programs and contractile function of borderzone fibroblasts, and increased vulnerability to pathological remodelling. In mice that died after myocardial infarction, IFNIC colonies were immediately adjacent to sites of ventricular rupture, while mice lacking IFNICs were protected from rupture and exhibited improved survival3. Together, these results reveal a pathological borderzone niche characterized by a cardiomyocyte-initiated innate immune response. We suggest that selective inhibition of IRF3 activation in non-immune cells could limit ischaemic cardiomyopathy while avoiding broad immunosuppression.
梗塞心臓の単一細胞および空間トランスクリプトーム解析により、機械的不安定化に応答する境界域の動的発生が明らかになった Single-cell and spatial transcriptomics of the infarcted heart define the dynamic onset of the border zone in response to mechanical destabilization
D. M. Calcagno,N. Taghdiri,V. K. Ninh,J. M. Mesfin,A. Toomu,R. Sehgal,J. Lee,Y. Liang,J. M. Duran,E. Adler,K. L. Christman,K. Zhang,F. Sheikh,Z. Fu & K. R. King
Nature Cardiovascular Research Published:17 November 2022
DOI:https://doi.org/10.1038/s44161-022-00160-3
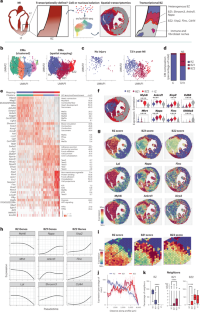
Abstract
The border zone (BZ) of the infarcted heart is a geographically complex and biologically enigmatic interface separating poorly perfused infarct zones (IZs) from remote zones (RZs). The cellular and molecular mechanisms of myocardial BZs are not well understood because microdissection inevitably combines them with uncontrolled amounts of RZs and IZs. Here, we use single-cell/nucleus RNA sequencing, spatial transcriptomics and multiplexed RNA fluorescence in situ hybridization to redefine the BZ based on cardiomyocyte transcriptomes. BZ1 (Nppa+Xirp2–) forms a hundreds-of-micrometer-thick layer of morphologically intact cells adjacent to RZs that are detectable within an hour of injury. Meanwhile, BZ2 (Nppa+Xirp2+) forms a near-single-cell-thick layer of morphologically distorted cardiomyocytes at the IZ edge that colocalize with matricellular protein-expressing myofibroblasts and express predominantly mechanotransduction genes. Surprisingly, mechanical injury alone is sufficient to induce BZ genes. We propose a ‘loss of neighbor’ hypothesis to explain how ischemic cell death mechanically destabilizes the BZ to induce its transcriptional response.
IRF3とI型インターフェロンが心筋梗塞に致命的な反応を引き起こす IRF3 and type I interferons fuel a fatal response to myocardial infarction
Kevin R King,Aaron D Aguirre,Yu-Xiang Ye,Yuan Sun,Jason D Roh,Richard P Ng Jr,Rainer H Kohler,Sean P Arlauckas,Yoshiko Iwamoto,Andrej Savol,Ruslan I Sadreyev,Mark Kelly,Timothy P Fitzgibbons,Katherine A Fitzgerald,Timothy Mitchison,Peter Libby,Matthias Nahrendorf & Ralph Weissleder
Nature Medicine Published:06 November 2017
DOI:https://doi.org/10.1038/nm.4428
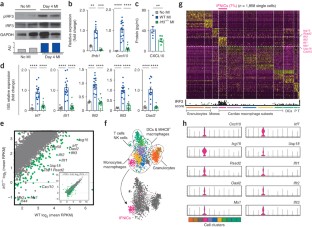
Abstract
Interferon regulatory factor 3 (IRF3) and type I interferons (IFNs) protect against infections1 and cancer2, but excessive IRF3 activation and type I IFN production cause autoinflammatory conditions such as Aicardi–Goutières syndrome3,4 and STING-associated vasculopathy of infancy (SAVI)3. Myocardial infarction (MI) elicits inflammation5, but the dominant molecular drivers of MI-associated inflammation remain unclear. Here we show that ischemic cell death and uptake of cell debris by macrophages in the heart fuel a fatal response to MI by activating IRF3 and type I IFN production. In mice, single-cell RNA-seq analysis of 4,215 leukocytes isolated from infarcted and non-infarcted hearts showed that MI provokes activation of an IRF3–interferon axis in a distinct population of interferon-inducible cells (IFNICs) that were classified as cardiac macrophages. Mice genetically deficient in cyclic GMP-AMP synthase (cGAS), its adaptor STING, IRF3, or the type I IFN receptor IFNAR exhibited impaired interferon-stimulated gene (ISG) expression and, in the case of mice deficient in IRF3 or IFNAR, improved survival after MI as compared to controls. Interruption of IRF3-dependent signaling resulted in decreased cardiac expression of inflammatory cytokines and chemokines and decreased inflammatory cell infiltration of the heart, as well as in attenuated ventricular dilation and improved cardiac function. Similarly, treatment of mice with an IFNAR-neutralizing antibody after MI ablated the interferon response and improved left ventricular dysfunction and survival. These results identify IRF3 and the type I IFN response as a potential therapeutic target for post-MI cardioprotection.
