
2025-04-09 カリフォルニア大学サンディエゴ校(UCSD)
<関連情報>
- https://today.ucsd.edu/story/a-comprehensive-map-of-the-human-cell
- https://www.nature.com/articles/s41586-025-08878-3
- https://www.nature.com/articles/s41592-024-02525-x
構造・機能ゲノミクスの基盤としてのマルチモーダルセルマップ Multimodal cell maps as a foundation for structural and functional genomics
Leah V. Schaffer,Mengzhou Hu,Gege Qian,Kyung-Mee Moon,Abantika Pal,Neelesh Soni,Andrew P. Latham,Laura Pontano Vaites,Dorothy Tsai,Nicole M. Mattson,Katherine Licon,Robin Bachelder,Anthony Cesnik,Ishan Gaur,Trang Le,William Leineweber,Aji Palar,Ernst Pulido,Yue Qin,Xiaoyu Zhao,Christopher Churas,Joanna Lenkiewicz,Jing Chen,Keiichiro Ono,… Trey Ideker
Nature Published:09 April 2025
DOI:https://doi.org/10.1038/s41586-025-08878-3
")
Abstract
Human cells consist of a complex hierarchy of components, many of which remain unexplored. Here we construct a global map of human subcellular architecture through joint measurement of biophysical interactions and immunofluorescence images for over 5,100 proteins in U2OS osteosarcoma cells. Self-supervised multimodal data integration resolves 275 molecular assemblies spanning the range of 10-8 to 10-5 m, which we validate systematically using whole-cell size-exclusion chromatography and annotate using large language models. We explore key applications in structural biology, yielding structures for 111 heterodimeric complexes and an expanded Rag–Ragulator assembly. The map assigns unexpected functions to 975 proteins, including roles for C18orf21 in RNA processing and DPP9 in interferon signalling, and identifies assemblies with multiple localizations or cell type specificity. It decodes paediatric cancer genomes4, identifying 21 recurrently mutated assemblies and implicating 102 validated new cancer proteins. The associated Cell Visualization Portal and Mapping Toolkit provide a reference platform for structural and functional cell biology.
遺伝子セット機能発見のための大規模言語モデルの評価 Evaluation of large language models for discovery of gene set function
Mengzhou Hu,Sahar Alkhairy,Ingoo Lee,Rudolf T. Pillich,Dylan Fong,Kevin Smith,Robin Bachelder,Trey Ideker & Dexter Pratt
Nature Methods Published:28 November 2024
DOI:https://doi.org/10.1038/s41592-024-02525-x
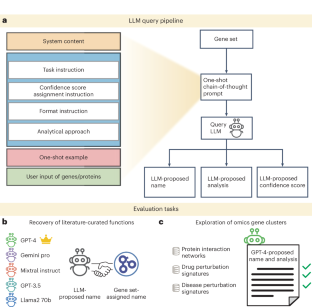
Abstract
Gene set enrichment is a mainstay of functional genomics, but it relies on gene function databases that are incomplete. Here we evaluate five large language models (LLMs) for their ability to discover the common functions represented by a gene set, supported by molecular rationale and a self-confidence assessment. For curated gene sets from Gene Ontology, GPT-4 suggests functions similar to the curated name in 73% of cases, with higher self-confidence predicting higher similarity. Conversely, random gene sets correctly yield zero confidence in 87% of cases. Other LLMs (GPT-3.5, Gemini Pro, Mixtral Instruct and Llama2 70b) vary in function recovery but are falsely confident for random sets. In gene clusters from omics data, GPT-4 identifies common functions for 45% of cases, fewer than functional enrichment but with higher specificity and gene coverage. Manual review of supporting rationale and citations finds these functions are largely verifiable. These results position LLMs as valuable omics assistants.
")
")