
")
2025-05-14 ニューヨーク大学(NYU)

Corn growing in the Irene Rose Sohn Zegar Memorial Greenhouse on the top floor of NYU’s Center for Genomics and Systems Biology. Credit: Tracey Friedman/NYU
<関連情報>
- https://www.nyu.edu/about/news-publications/news/2025/may/ai-nue-regulons.html
- https://academic.oup.com/plcell/advance-article/doi/10.1093/plcell/koaf093/8129742
- https://www.nature.com/articles/s41467-021-25893-w
モデルから作物へ保存されたNUEレギュロンが、窒素利用効率の機械学習による予測を強化する NUE regulons conserved model-to-crop enhance machine learning predictions of nitrogen use efficiency
Ji Huang , Chia-Yi Cheng , Matthew D Brooks , Tim L Jeffers , Nathan M Doner , Hung-Jui Shih , Samantha Frangos , Manpreet Singh Katari , Gloria M Coruzzi
The Plant Cell Published:14 May 2025
DOI:https://doi.org/10.1093/plcell/koaf093
Abstract
Systems biology aims to uncover gene regulatory networks (GRNs) for agricultural traits, but validating them in crops is challenging. We addressed this challenge by learning and validating model-to-crop GRN regulons governing nitrogen use efficiency (NUE). First, a fine-scale time-course nitrogen (N) response transcriptome analysis revealed a conserved temporal N response cascade in maize (Zea mays) and Arabidopsis (Arabidopsis thaliana). This data was used to infer time-based causal transcription factor (TF) target edges in N-regulated GRNs (N-GRNs). By validating 23 maize TFs in a cell-based TF-perturbation assay (TARGET), precision/recall analysis enabled us to prune high-confidence edges between ∼200 TFs/700 maize target genes. We next learned gene-to-NUE trait scores using XGBoost machine learning models trained on conserved N-responsive genes across maize and Arabidopsis accessions. By integrating NUE gene scores within our N-GRN, we ranked maize TFs based on a cumulative NUE regulon score. Regulons for top-ranked TFs were validated using the cell-based TARGET assay in maize (e.g. ZmMYB34/R3→24 targets) and the Arabidopsis ZmMYB34/R3 ortholog (e.g. AtDIV1→23 targets). The genes in this NUE regulon significantly enhanced the ability of XGBoost models to predict NUE traits in both maize and Arabidopsis. Thus, our pipeline for identifying NUE regulons that combines GRN inference, machine learning, and orthologous network regulons offers a strategic framework for crop trait improvement.
進化的情報に基づく機械学習が遺伝子と表現型の関係を予測する力を高める Evolutionarily informed machine learning enhances the power of predictive gene-to-phenotype relationships
Chia-Yi Cheng,Ying Li,Kranthi Varala,Jessica Bubert,Ji Huang,Grace J. Kim,Justin Halim,Jennifer Arp,Hung-Jui S. Shih,Grace Levinson,Seo Hyun Park,Ha Young Cho,Stephen P. Moose & Gloria M. Coruzzi
Nature Communications Published:24 September 2021
DOI:https://doi.org/10.1038/s41467-021-25893-w
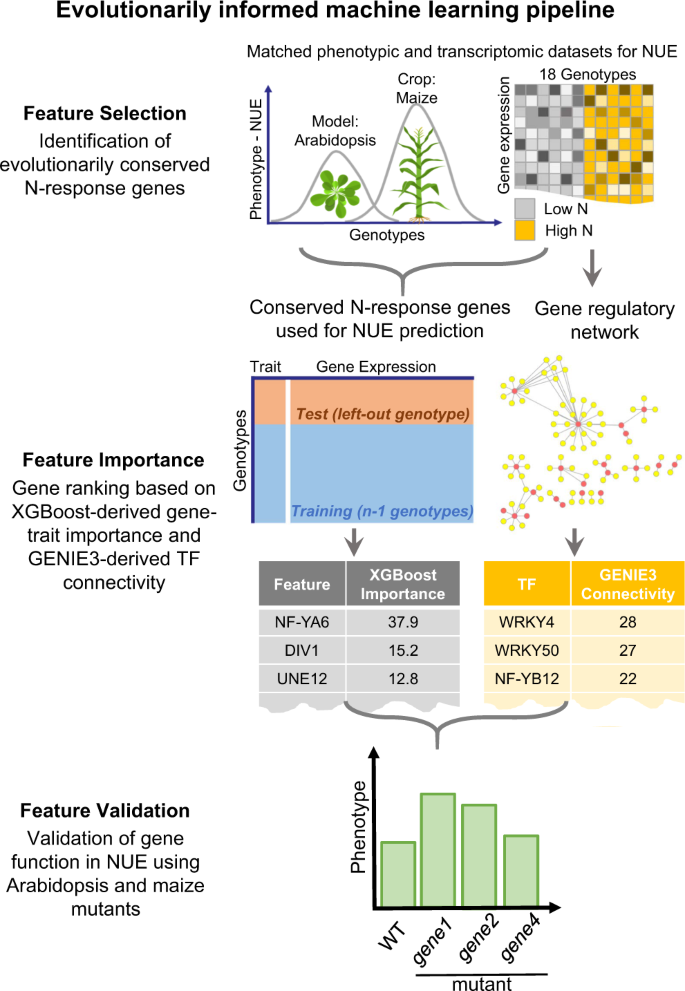
Abstract
Inferring phenotypic outcomes from genomic features is both a promise and challenge for systems biology. Using gene expression data to predict phenotypic outcomes, and functionally validating the genes with predictive powers are two challenges we address in this study. We applied an evolutionarily informed machine learning approach to predict phenotypes based on transcriptome responses shared both within and across species. Specifically, we exploited the phenotypic diversity in nitrogen use efficiency and evolutionarily conserved transcriptome responses to nitrogen treatments across Arabidopsis accessions and maize varieties. We demonstrate that using evolutionarily conserved nitrogen responsive genes is a biologically principled approach to reduce the feature dimensionality in machine learning that ultimately improved the predictive power of our gene-to-trait models. Further, we functionally validated seven candidate transcription factors with predictive power for NUE outcomes in Arabidopsis and one in maize. Moreover, application of our evolutionarily informed pipeline to other species including rice and mice models underscores its potential to uncover genes affecting any physiological or clinical traits of interest across biology, agriculture, or medicine.
")
")