2021-07-09 理化学研究所
理化学研究所(理研)生命機能科学研究センター分子機能シミュレーションチームの杉田有治チームリーダー(開拓研究本部杉田理論分子科学研究室主任研究員、計算科学研究センター粒子系生物物理研究チームチームリーダー)、笠原健人研究員(研究当時)、タンパク質機能・構造研究チームの白水美香子チームリーダーらの国際共同研究グループは、現実の細胞内に近い分子混雑環境[1]での酵素とその阻害剤の結合過程を、分子動力学(MD)計算[2]によりシミュレーションすることに成功しました。
本研究から明らかになった、分子混雑環境で阻害剤の効果が低下する機構や、希薄水溶液中とは異なる結合経路[3]の存在は、今後、生体内環境を考慮した新たなイン・シリコ創薬[4]の進展に貢献すると期待できます。
創薬の基本戦略の一つは、疾患に関わるタンパク質(酵素)の活性を阻害する薬剤基質(阻害剤)の探索です。阻害剤は酵素と結合して作用しますが、さまざまなタンパク質や核酸、代謝物などが高濃度に存在する細胞内環境で、酵素と阻害剤がどのように結合するのかはよく分かっていませんでした。
今回、国際共同研究グループは、スーパーコンピュータ「京」[5]と米国のMD専用計算機「Anton2」[6]を用いて、がんに関わる酵素とその阻害剤、およびそれらを囲むタンパク質(混雑タンパク質[1])の相互作用をMDシミュレーションで解析しました。その結果、混雑タンパク質が阻害剤を吸着することで、酵素周囲の阻害剤の実効濃度が減少し、酵素活性阻害の効率が低下することが分かりました。さらに、阻害剤が酵素に接近する初期過程も変化し、希薄水溶液中とは異なる経路をたどって阻害剤が結合ポケット[7]に挿入されることを発見しました。
本研究は、オンライン科学雑誌『Nature Communications』(7月2日付)に掲載されました。

分子(タンパク質)混雑環境における酵素(c-Srcキナーゼ)と阻害剤の結合
背景
細胞の増殖など生命機能の多くは、生体内の反応に必須なタンパク質である酵素が、基質分子と結合することで発現します。従って、その機能が異常化したとき、腫瘍形成などの重篤な疾患の発症に至ります。近年の創薬では、異常化した酵素の機能を阻害する基質分子(阻害剤)を効率よく設計することで、従来は治療が困難な疾患に対する薬剤を探索・開発し、健康・長寿社会の実現への貢献を目指しています。
構造解析技術[8]の進歩に伴い、タンパク質およびタンパク質と基質分子の複合体の立体構造情報は急速に蓄積されつつあります。しかし、タンパク質や酵素が働く現場である細胞内環境で、実際にこれらがどのような立体構造をとっているのか、タンパク質と基質分子の間に働く力(分子間相互作用)はどうなっているのか、それらの分子がどのように動いているのか(ダイナミクス)、といった情報を得るための実験手法は限られていました。特に、細胞内環境は多くのタンパク質や核酸、代謝物などが高濃度に存在する「分子混雑環境」であることが知られています。このような複雑な環境で、酵素と基質分子の間にどのような力が働いているのか、どのようなプロセスで結合するのかなどの詳細はよく分かっていませんでした。
分子動力学(MD)計算に代表される分子シミュレーションは、実験で直接観測することが難しいタンパク質など生体分子の動きをコンピュータ上で再現し、原子レベルで解析する手法です。近年のスーパーコンピュータの飛躍的な進歩に伴って急速な進歩を遂げており、酵素と基質分子の結合過程などを正確に予測することも可能になってきています。
研究手法と成果
国際共同研究グループは、分子混雑環境での酵素と阻害剤の結合モデルとして、細胞のがん化に関与するタンパク質リン酸化酵素の一つであるc-Srcキナーゼ[9]とATP競合性阻害剤[10]の結合に着目し、希薄水溶液中と分子混雑環境の二つの条件で長時間のMD計算を行いました。分子混雑環境条件としては、c-SrcキナーゼやATP競合性阻害剤と直接相互作用しないと考えられているウシ血清アルブミン(Bovine Serum Albumin、BSA)を混雑タンパク質として含めたシミュレーションを行いました。
本研究では、理研を中心に開発している超並列MDソフトウェアであるGENESIS(GENeralized Ensemble SImulation System)[11]を、スーパーコンピュータ「京」上で用いることにより、濃度の異なる複数のタンパク質混雑環境でシミュレーションを行いました。さらに、米国のD.E.Shawらのグループによって開発されたMD計算に特化した専用計算機である「Anton2」を併用することで、長時間のシミュレーションも実現しました。この二つの計算資源を用いて、合計で約100マイクロ秒(1マイクロ秒は1,000分の1ミリ秒)間にわたる結合過程のシミュレーション計算を行うことに成功しました。タンパク質と基質分子の動的な相互作用はミリ秒までのスケールで起きると考えられており、今回のシミュレーション(約0.1ミリ秒)はそれに迫るものです。
希薄環境中のシミュレーションでは、c-Srcキナーゼと複数の阻害剤が含まれていることから、阻害剤は拡散によって、容易にc-Srcキナーゼ表面に接近することができます(図1a)。一方、分子混雑環境のシミュレーションではc-Srcキナーゼと複数のBSAが存在するため、阻害剤は混雑タンパク質であるBSA表面に弱く吸着し、c-Srcキナーゼへの接近が妨げられます。このことは、BSAの濃度を段階的に上げていくと、c-Srcキナーゼ表面での阻害剤の存在確率が減少していき、逆にBSA表面上での阻害剤の存在確率が高まることで直接的に示されました(図1a)。
また、c-Srcキナーゼの表面には結合ポケット以外にも阻害剤が結合するサイトが複数存在することが分かっていますが、このような分子結合サイトのほとんどでは、BSAによる立体障害のため阻害剤の結合が妨げられました。ただしc-Srcキナーゼの結合ポケットには、希薄水溶液中および分子混雑環境のいずれのシミュレーションにおいても、阻害剤の結合が確認できました(図1b)。これは結合ポケットがc-Srcキナーゼ表面のくぼんだ位置に存在するため、BSA存在下でも結合サイトとして有効に機能していることを示しています。
なお、これらの計算結果はc-Srcキナーゼ、混雑タンパク質BSA、阻害剤などを含む溶液中での生化学実験を行うことによっても、実験的に検証することができました。すなわち、混雑タンパク質BSAの濃度を実験的に上げていくにつれて、酵素であるc-Srcキナーゼの酵素活性に対する阻害剤の効き目が低下する様子が明らかになりました。
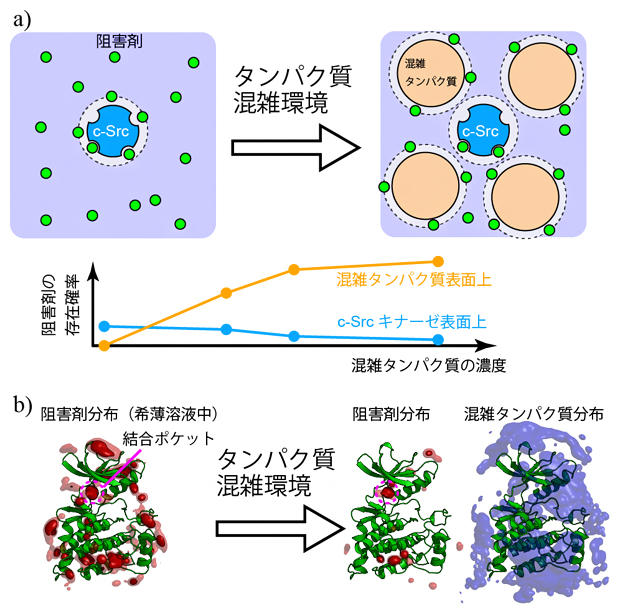
図1 溶液の混雑化がもたらすc-Srcキナーゼと阻害剤の結合の変化
a)酵素(c-Src)と阻害剤(緑)のみの希薄水溶液(左上)と、混雑タンパク質を加えた分子混雑環境の模式図(右上)。酵素一つに対して四つのBSAを加えた場合。阻害剤はBSA表面に弱く吸着し、c-Srcキナーゼ表面上の存在確率が低下するシミュレーション結果が得られた(下グラフ)。
b)c-Srcキナーゼの分子構造(緑色のリボンモデル)と、阻害剤(赤)、BSA(シアン)の分布をシミュレーションした結果。希薄溶液中では、阻害剤は結合ポケットを含むc-Srcキナーゼ表面のさまざまな領域に結合する(左)。分子混雑環境では、阻害剤は結合ポケットに入り込んでいるが、それ以外の領域に結合しにくくなっていた。このとき混雑タンパク質であるBSA(シアン、ここでは8分子)はc-Srcを囲むように分布しており、阻害剤の結合に対する立体障害となっていることが示された(右)。
酵素に対する阻害剤の効果は、阻害剤と酵素の結合の強さだけではなく、阻害剤が酵素に取り付いてから結合ポケットにたどり着くまでにどのような経路を選択するかにも影響されることが分かっています。そこで、今回行ったMDシミュレーションにおいて、阻害剤がc-Srcキナーゼの結合ポケットに近づくプロセスを、希薄水溶液中および分子混雑環境下のそれぞれについて複数回観測しました。すると、興味深いことに、阻害剤が結合ポケットに近づく経路はこの二つの条件で大きく異なり、酵素に取り付く際の最初の場所も違っていました(図2)。この変化は、分子混雑環境において、c-Srcキナーゼの立体構造や分子運動が、希薄水溶液中とは少しだけ異なることで生じるものと考えられましたが、詳細な分子機構を理解するためにはさらなる計算と解析が必要です。
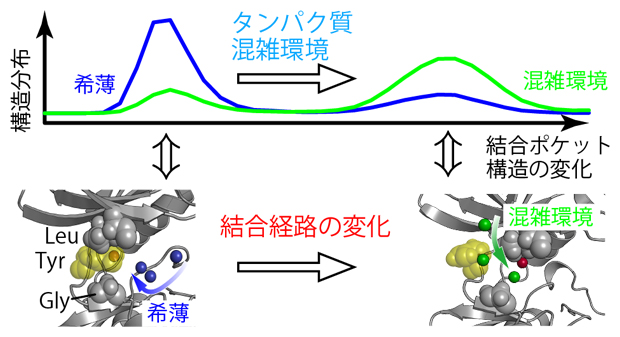
図2 溶液の混雑化に伴う結合ポケットの構造変化と結合経路の変化
上)c-Srcキナーゼは、希薄水溶液中と分子混雑環境のいずれにおいても、二つの異なる構造を示す。これらの構造の存在確率(構造分布)は、希薄水溶液中(青)と分子(タンパク質)混雑環境(緑)で逆転する。
下)希薄水溶液中と分子混雑環境でのc-Srcキナーゼの結合ポケット付近の分子構造と阻害剤の結合経路。希薄水溶液中では図の右側の方向から近づくのに対して(青矢印)、分子混雑環境のシミュレーションでは結合ポケットの上側から近づくことがほとんどであった(緑矢印)。
今後の期待
計算機を用いた効率的な薬剤開発の試みは近年非常に注目されており、多くのシミュレーション研究が行われるようになりました。しかし、そのほとんどは希薄水溶液中と同じ条件で計算されており、より細胞内環境に近いと考えられるタンパク質混雑環境の影響を考慮したシミュレーション研究は本研究が世界的にも初めての試みといえます。
新規薬剤の開発にかかる時間がさらに長期化している現在、本成果は、タンパク質混雑環境を考慮した新たな分子設計の可能性を示すものとして期待できます。さらに、本研究で用いたソフトウェアGENESISは、スーパーコンピュータ「富岳」[12]において高並列化効率でMD計算が実行できるようにチューニングされており、フリーソフトウェアとして公開されています。「富岳」などの優れた計算資源を有効に用いることで、細胞内環境をより深く理解し、生体内環境と近い条件での分子設計を実現できる日も近づいていると考えています。
補足説明
1.分子混雑環境、混雑タンパク質
細胞質中の生体高分子(主にタンパク質やRNA)の濃度は300~450mg/mlであり、多くの生体高分子が混み合った分子混雑環境である。このような細胞内分子混雑環境が生体高分子に果たす役割を調べるために、ターゲットタンパク質と直接反応しないと考えられる別のタンパク質を混雑物(混雑タンパク質)として含む実験・計算が行われている。
2.分子動力学(MD)計算
原子間に働く力を計算し、運動方程式を繰り返し解くことで、分子の動きを追跡する方法。MDはMolecular Dynamicsの略。
3.結合経路
阻害剤などの基質分子が標的タンパク質と出会うと、多くの場合、過渡的な結合と解離を繰り返しながら特定部位に移動し、安定な結合に至る。この軌跡を結合経路と呼ぶ。
4.イン・シリコ創薬
イン・シリコは「シリコン内で」を意味し、AIやバイオインフォマティクスなどコンピュータ技術を用いた創薬手法を表す。イン・シリコに対して、「生体内」にはイン・ビボ(in vivo)、「試験管内」にはイン・ビトロ(in vitro)という用語が用いられる。
5.スーパーコンピュータ「京」
文部科学省が推進する「革新的ハイパフォーマンス・コンピューティング・インフラ(HPCI)の構築」プログラムの中核システムとして、理研と富士通が共同で開発を行い、2012年に共用を開始した計算速度10ペタフロップス級のスーパーコンピュータ。2019年8月に運用終了。
6.MD専用計算機「Anton2」
米国D.E. Shawらのグループが開発する分子動力学(MD)計算の高速化に特化して設計されたスーパーコンピュータ。ピッツバーグスーパーコンピューティングセンターに設置されている。2009年に第一世代「ANTON」を発表、2014年に第二世代となる「ANTON2」を発表した。「ANTON2」は4X512ノードを搭載し大幅な高速化を実現している。
7.結合ポケット
原子レベルで見たタンパク質の表面には凹凸があり、その中でも低分子が結合しやすいポケット状のくぼんだ空間を結合ポケットと呼ぶ。
8.構造解析技術
X線結晶解析、核磁気共鳴(NMR)、クライオ電子顕微鏡法など、主にタンパク質を始めとする高分子の立体構造を解析する技術。
9.c-Src キナーゼ
Src遺伝子にコードされる非受容体型チロシンキナーゼタンパク質(他の分子のチロシン残基をリン酸化するタンパク質)。細胞成長の制御やがんと関係することが知られている。
10.ATP競合性阻害剤
本来の基質と競合的に酵素の基質結合部位である結合ポケットに入り込むことで、酵素反応を阻害する化合物を競合性阻害剤と呼ぶ。ATP競合性阻害剤は酵素との結合をめぐってATP(アデノシン三リン酸)と競合し、その酵素活性を阻害する低分子化合物のこと。
11.GENESIS
理研計算科学研究センターを中心に開発されている分子動力学ソフトウェア![]() 。細胞環境を含む大規模な生体分子系のシミュレーションやレプリカ交換法などの構造探索手法を利用できるという特徴を持つ。
。細胞環境を含む大規模な生体分子系のシミュレーションやレプリカ交換法などの構造探索手法を利用できるという特徴を持つ。
12.スーパーコンピュータ「富岳」
「京」の後継機。社会的・科学的課題の解決で日本の成長に貢献し、世界をリードする成果を生み出すことを目的とし、電力性能、計算性能、ユーザーの利便性・使い勝手の良さ、画期的な成果創出、ビッグデータやAI(人工知能)の加速機能の総合力において世界最高レベルのスーパーコンピュータ。15万8976個の中央演算装置(CPU)を搭載し、1秒間に約44京2010兆回の計算が可能。2020年6月と11月、2021年6月に世界のスパコンランキング「TOP500」「HPCG」「HPL-AI」「Graph500」で3期連続の世界一位を獲得した。
国際共同研究グループ
理化学研究所 生命機能科学研究センター
分子機能シミュレーションチーム
チームリーダー 杉田 有治(すぎた ゆうじ)
(開拓研究本部 杉田理論分子科学研究室 主任研究員、計算科学研究センター 粒子系生物物理研究 チームチームリーダー)
上級研究員(研究当時) 李 秀栄(り すよん)
(現 国立研究開発法人 医薬基盤・健康・栄養研究所)
研究員(研究当時) 笠原 健人(かさはら けんと)
(現 大阪大学 基礎工学部)
研究員 尾嶋 拓(おしま ひらく)
タンパク質機能・構造研究チーム
チームリーダー 白水 美香子(しろうず みかこ)
上級研究員 新野 睦子(にいの むつこ)
技師(研究当時) 津曲 千恵美(つまがり ちえみ)
テクニカルスタッフ 矢吹 有香子(やぶき ゆかこ)
ミシガン州立大学 生化学および分子生物学部
教授 マイケル・ファイグ(Michael Feig)
博士研究員(研究当時) ジェゴルシュ・ナブロツキ(Grzegorz Nawrocki)
前橋工科大学 工学部生命情報学科
准教授 優 乙石(ゆう いっせき)
研究支援
本研究は、理化学研究所運営費交付金(生命機能科学研究、理化学研究所新領域開拓課題(Dynamic Structural Biology, Biology of Intracellular Environments))で実施し、ポスト「京」重点課題1、「富岳」成果創出加速プログラム、科学研究費補助金基盤研究(S)「マルチスケール分子動力学シミュレーションによる細胞内分子動態の解明(研究代表者:杉田有治)」、米国NSF(MCB1817307)、米国NIH(R35 GM126948)による支援を受けて行われました。分子動力学シミュレーションは、理研HOKUSAI GreatWave/BigWaterfall、「京」(課題番号hp170254, hp180201, hp180274, hp190097, hp190181, hp200129, hp200135)、ピッツバーグスーパーコンピューティングセンターAnton2(PSCA18053P)の計算資源の提供を受け、実施しました。
原論文情報
Kento Kasahara, Suyong Re, Grzegorz Nawrocki, Hiraku Oshima, Chiemi Mishima-Tsumagari, Yukako Miyata-Yabuki, Mutsuko Kukimoto-Niino, Isseki Yu, Mikako Shirouzu, Michael Feig, Yuji Sugita, “Reduced Efficacy of a Src Kinase Inhibitor in Crowded Protein Solution”, Nature Communications, 10.1038/s41467-021-24349-5
発表者
理化学研究所
生命機能科学研究センター 分子機能シミュレーション研究チーム
チームリーダー 杉田 有治(すぎた ゆうじ)
(開拓研究本部 杉田理論分子科学研究室 主任研究員、計算科学研究センター
粒子系生物物理研究チーム チームリーダー)
研究員(研究当時) 笠原 健人(かさはら けんと)
タンパク質機能・構造研究チーム
チームリーダー 白水 美香子(しろうず みかこ)
報道担当
理化学研究所 広報室 報道担当

