
2023-06-27 国立がん研究センター
発表のポイント
- 腫瘍と対照正常組織のロングリードシークエンスデータをペアで扱い、高精度に後天的構造異常の検出を行うソフトウェア「nanomonsv」を世界に先駆けて開発し、広く活用されています。
- ショートリード・ロングリードの包括的な比較検討により、約95%という高い精度、かつ高感度な検出を達成しました。
- ロングリードシークエンスの特性を活用し、消化器系のがんや肺がんで多く見られる構造異常の一種である「モバイルエレメント挿入」を高感度に検出しました。
- 「Single breakend SVモジュール」というアルゴリズムを開発し、これまでの技術では検出不能であったセントロメアやテロメア配列を巻き込む構造異常の検出が可能であることを実証しました。
- 今後がんにおいてもロングリードシークエンスデータを使ったより精密な全ゲノム解析の重要性が増してきています。本研究成果は今後のゲノム情報解析の基盤となり、研究・医療に展開されることが期待されます。
概要
国立研究開発法人国立がん研究センター(理事長:中釜 斉、東京都中央区) 研究所(所長:間野博行)ゲノム解析基盤開発分野白石友一分野長らの研究グループは、同研究所分子腫瘍学分野(分野長:片岡圭亮)などとの共同研究により、ロングリードシークエンスデータからがんゲノムに生じている後天的構造異常を高精度に検出するソフトウェア「nanomonsv」の開発に成功しました。
ロングリードシークエンス技術の発展により、これまで主流となっているショートリードに比べて、高感度に後天的構造異常の検出が可能になると期待されております。一方で、腫瘍と対照正常組織の双方のロングリードシークエンスデータをペアで利用して、高精度に後天的構造異常の検出を達成できるソフトウェアの開発は、世界的にもほとんど実現されておりませんでした。
本研究では、開発したソフトウェアの包括的な比較検証を行い、ショートリードで検出可能な構造異常の多くがnanomonsvにより検出できることを明らかにしました。さらに、PCRによる検証から、約95%という高い精度、かつ高感度な検出ができていることを確認しました。また、消化器系のがんや肺がんで多く見られる「モバイルエレメント挿入」などの構造異常や、セントロメアやテロメア配列を巻き込む構造異常など、これまでのソフトウェアでは検出ができなかったタイプの構造異常を検出可能であることを実証しました。
今後がんにおいてもロングリードシークエンスデータを使ったより精密な全ゲノム解析の重要性が増してきています。本研究成果は今後のゲノム情報解析の基盤となり、研究・医療に展開されることが期待されます。
本研究成果は、英国科学誌「Nucleic Acids Research」に2023年6月20日に公開されました。
背景
がん細胞に特異的に生じている遺伝子変異のうち、大域的なゲノムの変化(後天的構造異常:一般的には50塩基以上で、欠失・挿入・重複・逆位・転座など)は、がんの発生・進展に非常に重要な役割を占めていると考えられてきました。シークエンス技術の革新により、原理的にはがんゲノムに生じている後天的構造異常をスクリーニングすることが可能となりました。一方で、ヒトゲノムの配列上には、いわゆるリピート領域が広く分布しており、これらの領域は、現在主流となっているDNAを断片化して、その両端の短い配列を解読するショートリード解析の対象外となっています(図1)。そのため、現在得られている後天的構造異常のリストはヒトゲノムの領域を完全に網羅したものではなく、ゲノム医療において治療標的になる後天的構造異常が十分に検出されていないと考えられてきました。
最近のロングリードシーケンシング技術の進歩により長いDNA(数万塩基単位)をそのまま解読することができるため、これまでより格段に広い領域の解析が可能となり、精緻な後天的構造異常のリストを得られることが期待されています。しかしながら、生殖細胞系列の構造異常を検出するソフトウェアは数多く開発されているものの、後天的構造異常、特に、腫瘍と対照正常のペアのシークエンスデータを用いた検出に対応するソフトウェアはほとんど開発がなされていませんでした。
研究成果
白石友一分野長らの研究グループは、腫瘍と対照正常のペアになったロングリードシークエンスデータから高精度に後天的構造異常を特定するソフトウェア「nanomonsv」を開発しました。GitHub上でオープンソースとして公開されており、以下のURLからアクセスが可能です。
nanomonsvのウェブサイト
1.約95%という高い精度
まず、3つの細胞株(COLO829, H2009, HCC1954)とその対照正常のロングリードデータ・高深度ショートリードデータを使って、精度検証を進めました。高深度ショートリードにおける5つの構造異常検出ツールの包括的な比較解析により、ショートリードシークエンスで検出される後天的構造異常の大部分は、ロングリードデータをnanomonsvで解析することで検出可能であることを示しました。PCRによる検証から、正答率は約95%と推測されました。また、約7~12%の後天的構造変異が新しく検出されました。
2.消化器系のがんや肺がんで多く見られる構造異常「モバイルエレメント挿入」を高感度に検出
構造変異の一部に、「モバイルエレメント挿入」と呼ばれる、DNAの一部(移動遺伝子要素)が一箇所から切り離され、DNAの別の場所に挿入される現象があります(図2)。こうしたモバイルエレメント挿入は、特に消化器系のがん、肺がんなどで多く見られることが知られています。本研究では、こうしたモバイルエレメント挿入がnanomonsvにより高感度で検出できることを示しました。さらにモバイルエレメント挿入を詳細に分類・特徴づけるための解析プログラムを作成し、挿入配列のゲノム起源の位置など、種々の特徴を得ることを示しました。
3.繰り返しの多いセントロメア・テロメア配列を巻き込む構造異常を検出
構造変異は通常、二つのゲノム上の切断点によって定義されます。しかし、セントロメアなどのように明確に定義されていない領域が多く存在するため、二つの切断点を共に特定することは難しいことがあります。「一つの切断点のみを特定できれば十分である」という原理に基づくアルゴリズム「Single Breakend SVモジュール(図3右)」を開発し、nanomonsvに搭載しています。3つの細胞株から合計91の単一切断点構造異常を特定することができました。顕著な例としては、著名ながん抑制遺伝子におけるRB1遺伝子に影響を与えるもので、2つの断片の逆位の後に、RB1の配列がセントロメアの配列に接続されている現象を同定しました(図4)。さらに、テロメア配列につながる単一切断点構造異常を複数個特定することができました。一つの例としては、X染色体において突然の切断点があり、約1,200のテロメア配列(TTAGGGの繰り返し)に繋がり、14番染色体の端に繋がるというものです(図5)。
これらのセントロメア・テロメア配列を巻き込む構造異常は他の構造異常検出ツールにも見逃されていましたが、このようなクラスの構造異常を検出が可能になることで、がんゲノムの進展のさらなる理解に役立つと期待されます。

図1:ショートリードとロングリードの違い
ショートリードでは、ゲノムに散りばめられているリピート領域(ピンクで示す)へのマッピングができず、リピート領域における変異は十分に検出できていなかった。ロングリードにより、数万塩基と長い塩基配列を解読することができ、染色体の構造変異の検出が容易になった。また、多くのリピート領域へのマッピングが可能になり、広い領域での解析が可能になった。一方で、ロングリードシークエンスで取得できる配列長よりも長いリピート領域(セントロメア配列など)(緑で示す)については、ロングリードシークエンスでもマッピングができず、解析のためにはさらに特別な方法論が必要である。
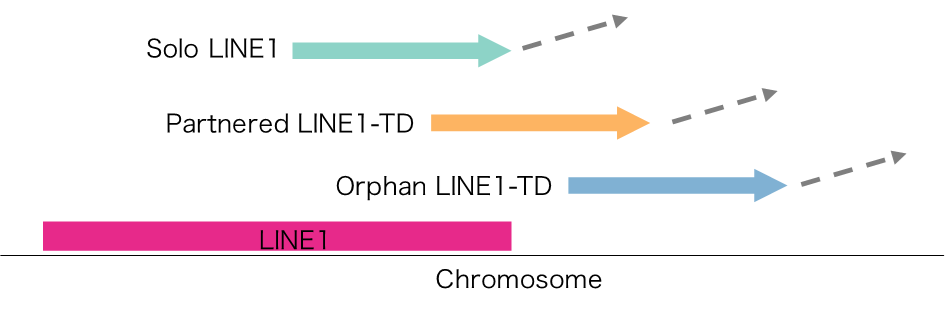
図2:代表的なモバイルエレメントであるLINE1挿入の分類
LINE1配列(+その下流配列)が転写されてから逆転写により別のゲノム位置に入る前に5’側が欠ける。最終的な挿入配列がLINE1配列自身、LINE1配列+下流配列、下流配列のみにより、Solo LINE1, Partnered LINE1-Transduction (TD), Orphan LINE1-TDに分類される。
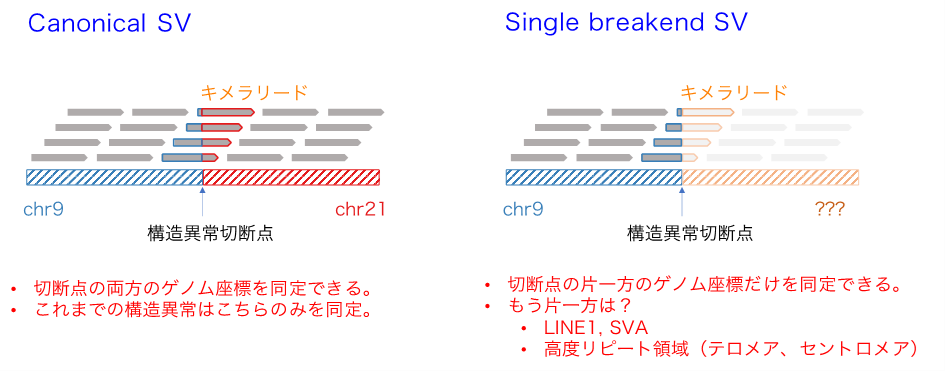
図3: 一般的な構造異常(Canonical SV)、切断点が一つだけ同定される構造異常(Single breakend SV)の違いの概念図
Single breakend SVモジュールは右のタイプの構造異常検出を達成する。
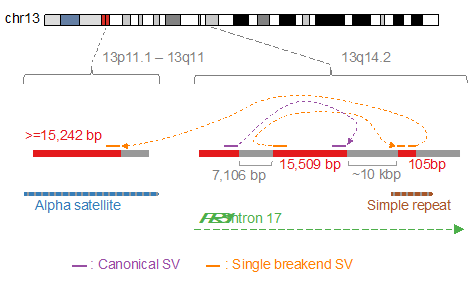
図4:RB1遺伝子における、セントロメアに繋がる単一切断点構造異常
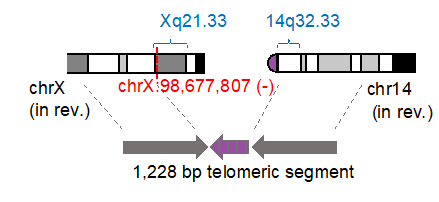
図5:テロメア配列を介した転座
展望
今回開発した「nanomonsv」は世界中の研究者にすでに利用されており、ゲノム解析におけるロングリードシークエンス技術の発展の礎となっています。今後開発した技術を基盤に、世界中の研究者による技術革新が始まっており、さらなる計測技術・情報解析技術の革新がもたらされることが期待されています。
近年のヒトゲノム完全長解読 (telomere-to-telomere)コンソーシアムにより、セントロメア・テロメア領域などの難読領域を含めて、ヒト一人のゲノム塩基配列の完全な解読がなされました。こうした動きはやがてがんゲノム領域にも波及し、近い将来我々は完全ながんゲノム配列を得て、それを元に研究・医療が展開される状況になることが予想されます。
今回の研究ではSingle breakend SVモジュールにより、切断点の片側がセントロメア・テロメア領域などの難読領域に位置する後天的構造異常の検出ができることを示しました。一方で、両方の切断点が難読領域に位置している場合には、現在のアルゴリズムでも検出ができません。今後、さらに難読領域を含めた後天的構造異常のプロファイルを得て、完全ながんゲノムの再構成を行うためには、ヒトゲノム完全長解読コンソーシアムを中心に開発されている技術を使って、個別の患者の正常のゲノムを完全に決定し、その上での一連の解析を展開することを考えています。また、ヒト・パンゲノム・リファレンス・コンソーシアム(Human Pangenome Reference Consortium)で構築が進んでいる、ヒトゲノムの集団・個体間のバリエーションを組み込んだグラフゲノムを上手く利用する方法論の開発も重要です。ロングリードシークエンスの持つポテンシャルを最大限に引き出して、がんゲノムの理解に繋げるための情報解析手法の開発をさらに推進したいと考えています。
論文情報
雑誌名
Nucleic Acids Research
タイトル
Precise characterization of somatic complex structural variations from tumor/control paired long-read sequencing data with nanomonsv
著者
Yuichi Shiraishi, Junji Koya, Kenichi Chiba, Ai Okada, Yasuhito Arai, Yuki Saito, Tatsuhiro Shibata, Keisuke Kataoka
DOI
10.1093/nar/gkad526/7201946
URL
https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkad526/7201946
掲載日
2023年6月20日
研究費
- 国立研究開発法人日本医療研究開発機構(AMED) 次世代がん医療創生研究事業 「新規検出アルゴリズムとロングリードシーケンスを併用した非古典的構造異常の全がん解析」 (代表: 白石友一)
- 国立研究開発法人日本医療研究開発機構(AMED) 難治性疾患実用化研究事業 「長鎖・短差鎖シークエンス技術の統合による構造変異の検出と非翻訳領域情報を駆使した未診断症例の解決」 (代表: 小崎健次郎)
- 国立がん研究センター研究開発費(2021-A-3)「長鎖シークエンスを用いた研究基盤の構築と臨床的有用性の検証」(代表: 白石友一)
問い合わせ先
研究に関する問い合わせ
国立研究開発法人国立がん研究センター
研究所 ゲノム解析基盤開発分野 白石友一
広報窓口
国立がん研究センター
企画戦略局 広報企画室
")
