
2024-03-05 国立がん研究センター,理化学研究所
発表のポイント
- 網羅的DNAメチル化データ解析において、データセット・プラットフォームによるバイアスによって横断的解析が困難という課題がありました。
- 研究グループは、機械学習技術の一つである非負値行列因子分解(NMF)を用いて、網羅的DNAメチル化データのための新たなデータ解析手法を開発しました。
- 新たに開発したデータ解析手法は、複数のデータセットを統合して解析することを容易にし、希少がんなど、単一施設で収集できる症例の数に限りのある疾患のデータ分析が促進されることが期待できます。
- 本研究成果により、がんのエピジェネティックな機序解明や創薬標的探索が進むことが期待されます。
概要
国立研究開発法人国立がん研究センター(理事長:中釜 斉、東京都中央区)研究所(所長:間野博行)医療AI研究開発分野の高澤建・外来研究員(理化学研究所革新知能統合研究センター研究員)、浜本隆二分野長などからなる研究グループは、機械学習技術の一つである非負値行列因子分解(NMF)注1を用いた網羅的DNAメチル化注2データのための新たなデータ解析手法「methPLIER(メスプライヤー)」を開発しました。
生物の発生や細胞分化の過程で動的に変化するDNAメチル化については、これまで多くの研究がなされ、がん研究領域においても、がん関連遺伝子の発現制御との関連など重要な報告がされています。そのため、さらなる研究の推進が求められていますが、従来のDNAメチル化分析ツールは、一つのデータセット内の比較分析に焦点を当てていたため、異なるデータセット間の比較や、希少疾患研究、異なる機関間の研究などを行うのが難しい状況でした。今回研究グループが開発したmethPLIERは、サンプル間およびデータセット間の比較分析や、横断的なDNAメチル化データ分析が容易になり、DNAメチル化データリソースの利活用促進に寄与することが期待されます。
この研究成果は、国際学術雑誌「Experimental & Molecular Medicine」オンライン版(3月4日付)に掲載されました。
図1:本研究で構築したmethPLIERの概略図と、methPLIERの基本機能

網羅的DNAメチル化データを行列分解し、クラスタリング・生存時間解析・データの可視化を行うことが可能。
背景
DNAメチル化は細胞・組織ごとに特異的なパターンを形成しており、遺伝子発現の制御に関わっています。がんを含む様々な疾患においても、DNAメチル化パターンの変化によって、遺伝子発現の異常な活性・抑制が起こることが知られています。網羅的DNAメチル化解析注3の解析プラットフォームとして、BeadChip解析と呼ばれるIllumina HumanMethylation BeadChipや次世代シークエンサーを用いた全ゲノムバイサルファイトシークエンス(Whole Genome Bisulfite-sequencing: WGBS)などが開発されています。特にがん研究領域では、第2世代BeadChip解析のHumanMethylation 450(HM450)が広く使われており、Gene Expression Omnibus(GEO)やThe Cancer Genome Atlas(TCGA)などの研究用公共データベースに多くのHM450データが登録されています。このようなデータに蓄積されてきた網羅的DNAメチル化データを利活用することで、単一施設では収集可能症例数に限度のある希少がんの研究や、他の研究グループによるデータセットとの統合解析が可能となります。しかし、研究グループや解析プラットフォームが異なることによるデータ分布のバイアスにより、本来のデータに含まれる生物学的な特徴が捉えにくくなってしまう場合があり、統合解析の際の障壁となっていました。
研究成果
そこで本研究ではデータセットやプラットフォームの違いによるデータバイアスによる影響を低減させるための新たなDNAメチル化データ解析手法、methPLIERを開発しました。methPLIERは、Pathway Level Information Extractor(PLIER)と呼ばれる遺伝子発現解析手法を、網羅的DNAメチル化データ解析に応用した手法で、非負行列因子分解(NMF)と知識行列による正則化および転移学習を組み合わせたものです。
- 大規模データセットに対する非負値行列因子分解
研究室等で得られる小さなデータセットをそのまま解析してしまうと、データセット中に含まれる些細な違いや、ノイズに対してもデータセットの特徴として抽出してしまう可能性があります。そこでmethPLIERでは、TCGAから取得した9,756サンプルのDNAメチル化データを用いて、大規模データ中に含まれる潜在特徴(Latent variables: LVs)を予め抽出し、研究者が解析したい小さなデータセットにLVsがどの程度含まれているかを表現します。これにより、解析したいデータセットに含まれるノイズに解析結果が左右されにくくなり、異なるデータセット同士の統合解析が行いやすくなると考えられます。またLVsの含有量を表す際、「マイナス30%含まれている」というような負の値を用いた表現では解釈性が低下することから、LVsの含有量(因子負荷行列, 図1のB)が必ず正の値を取るような行列分解手法、NMFを用いています。 - 知識行列を用いた潜在特徴行列の正則化
一般にLVs行列をNMFで求める場合、得られるLVs行列が生物学的な意味を持つことが担保されていません。そこでmethPLIERでは、LVs行列の生物学的な解釈性を高めるために、キュレーション済みの遺伝子セットやパスウェイデータベースをもとに知識行列(図1のU)を作成し、LVs行列が知識行列をスパースに含むような正則化条件を加えています。これにより、解析したいサンプルに多く含まれるLVsがどのような生物学的特性をもっているのかが解釈しやすくなることが考えられます。
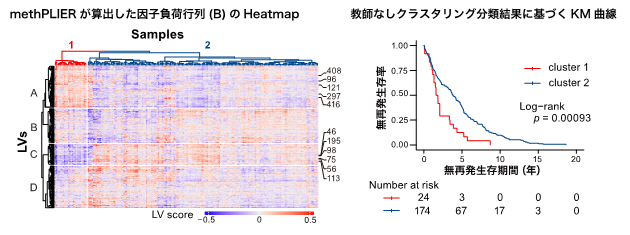
このようにして構築したmethPLIERを用いて、GEOに登録されている肺がんデータセットの解析を行いました(図2)。methPLIERを用いてデータセットから因子負荷行列を取得し、教師なしクラスタリング分類を行い2つのクラスタに分類しました。分類結果に基づき、無再発生存期間に対する生存時間分析を行ったところ、2群間において無再発生存時間に有意な差が認められました。このことから、methPLIERはデータセット内の生物学的特性を捉えることが可能な解析手法であることが示唆されました。
図2:肺がんデータセットに対するmethPLIERを用いた解析
データセット内の生物学的な特徴を捉えることが可能。
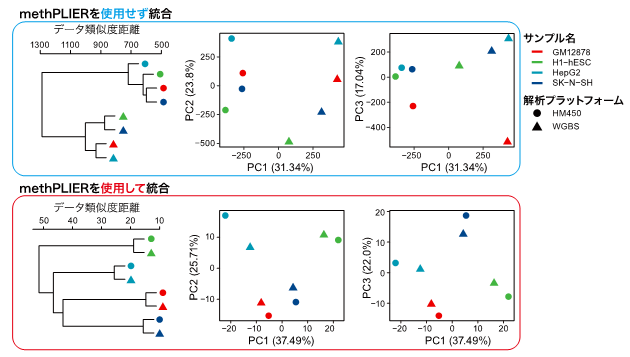
また、異なるプラットフォーム間のバイアスを低減できるかを確かめるために、同一細胞に対して異なるプラットフォームによって取得したDNAメチル化データを、methPLIERを用いずに統合した場合と用いた後に得られた因子負荷行列を統合したデータに対して教師なしクラスタリング分類を行い、データバイアスに対する低減効果を確認しました(図3)。methPLIERを用いずに統合した場合は、サンプルごとに分類されずにプラットフォームごとに分類されてしまいました。一方、methPILERを用いた後に統合した場合は、サンプルごとにデータが分類されており、プラットフォーム間のデータバイアスが低減されていることが示唆されました。図3:解析プラットフォームが異なるデータを統合した際の、教師なしクラスタリングの結果
methPLIERを用いることで、解析プラットフォームの違いによるバイアスが低減され、サンプルごとにクラスタが形成されている。
展望
本研究で構築したmethPILERは、データセットやプラットフォーム間のバイアスを低減させることでデータ横断的な統合解析を容易にします。今後、methPLIERを用いたデータセット横断的な解析が促進されることで、単一施設や研究機関で大規模なDNAメチル化データを収集することが困難な希少がんなどの研究が促進され、がんのエピジェネティックな機序解明や創薬標的探索が進むことが期待できます。
発表論文
雑誌名
Experimental & Molecular Medicine
タイトル
Advances in cancer DNA methylation analysis with methPLIER: use of non-negative matrix factorization and knowledge-based constraints to enhance biological interpretability
著者
Ken Takasawa, Ken Asada, Syuzo Kaneko, Kouya Shiraishi, Hidenori Machino, Satoshi Takahashi, Norio Shinkai, Nobuji Kouno, Kazuma Kobayashi, Masaaki Komatsu, Takaaki Mizuno, Yu Okubo, Masami Mukai, Tatsuya Yoshida, Yukihiro Yoshida, Hidehito Horinouchi, Shun-Ichi Watanabe, Yuichiro Ohe, Yasushi Yatabe, Takashi Kohno, Ryuji Hamamoto (*Corresponding Author)
DOI
10.1038/s12276-024-01173-7
掲載日
2024年3月4日付(オンライン・プレ・リリース)
URL
https://www.nature.com/articles/s12276-024-01173-7
研究費
- 科学技術振興機構(JST)・戦略的創造研究推進事業(JST CREST)「人工知能を用いた統合的ながん医療システムの開発」(研究代表者名: 浜本 隆二)
- 科学技術振興機構(JST)・AIPプロジェクト(AIP-PRISM)「人工知能技術を活用した革新的ながん創薬システムの開発」(研究代表者名: 浜本 隆二)
- 内閣府科学技術・イノベーション推進事務局 研究開発とSociety5.0との橋渡しプログラム(BRIDGE)「医療デジタルツインの発展に資するデジタル医療データバンク構想」(研究総括:浜本 隆二)
発表者
国立がん研究センター
研究所
医療AI研究開発分野:高澤建(理化学研究所革新知能統合研究センター研究員; 筆頭著者)、河野伸次、小林和馬、金子修三、浜本隆二(責任著者)
ゲノム生物学研究分野:白石航也、河野隆志
中央病院
呼吸器内科:水野孝昭、吉田達哉、堀之内秀仁、大江裕一郎
呼吸器外科:大久保祐、吉田幸弘、渡辺俊一
病理診断科:谷田部恭
医療情報部:向井まさみ
理化学研究所革新知能統合研究センター
がん探索医療研究チーム:新海典夫、町野英徳、高橋慧、浅田健、小松正明
用語説明
注1 非負値行列因子分解(NMF)
非負行列因子分解(Non-negative Matrix Factorization、NMF)は、データ分析やパターン認識などに使用される線形代数の技法です。この技法の主な目的は、非負のデータ行列を、同じく非負の2つの因子行列に分解することです。非負行列因子分解は、行列の各要素が非負である場合に特に有効です。
NMFの基本的な考え方は、与えられた非負のデータ行列V(例えば、サイズがm×n)を、2つの低ランクの非負行列 W (サイズが m×k)と H (サイズが k×n)に分解することです。ここで、k は V のランクよりも小さい値です。この分解により、元のデータ行列V は、これら2つの行列の積 WH によって近似されます。NMFの応用例としては、画像処理、テキストマイニング、音声認識、バイオインフォマティクスなどがあります。たとえば、画像処理においては、NMFを使用して画像を異なる部分やパターンに分解し、これらの特徴を分析することができます。テキストデータにおいては、異なるトピックやパターンを抽出するために使用されることもあります。NMFは、データの潜在的な構造を発見し、解釈可能な成分に分解することが可能であり、多くの実用的な応用において有用なツールとなっています。
注2 DNAメチル化
DNAを構成する塩基A(アデニン)、C(シトシン)、G(グアニン)、T(チミン)のうち、主にCとGが並ぶ部位(CpG)のCにメチル基(-CH3)が付くことをいいます。発生時期の細胞の種類の決定や遺伝子発現の制御などに関与しており、生活習慣や環境化学物質の曝露などによって後天的に変化します。
注3 網羅的DNAメチル化解析
網羅的DNAメチル化解析は、生物学や遺伝学において、遺伝子の発現を調節するエピジェネティックな修飾であるDNAメチル化の全体的なパターンを調査する手法です。DNAにメチル基が付加される現象をゲノム全体で詳細に分析することで、遺伝子の活性変化や疾患の進行に及ぼす影響を理解することができます。この解析は、Bisulfite-sequencingやメチル化DNA免疫沈降などの技術を用いて行われ、がんや神経疾患などの健康問題研究において重要な役割を果たしています。
お問い合わせ先
研究に関するお問合せ
国立研究開発法人国立がん研究センター研究所
医療AI研究開発分野 分野長 浜本 隆二
国立研究開発法人理化学研究所 革新知能統合研究センター
がん探索医療研究チーム 研究員 高澤 建
広報窓口
国立研究開発法人国立がん研究センター
企画戦略局 広報企画室
国立研究開発法人理化学研究所 広報室 報道担当

