
2018-03-13 理化学研究所,日本医療研究開発機構
要旨
理化学研究所(理研)情報基盤センターバイオインフォマティクス研究開発ユニットの笹川洋平上級センター研究員、團野宏樹センター研究員(研究当時)、二階堂愛ユニットリーダーらの共同研究チーム※は、大量の1細胞由来RNAを網羅的、高精度かつ低コストで計測する高出力型1細胞RNAシーケンス法「Quartz-Seq2(クォーツ・セックツー)」[1]を開発しました。
私たちの体は、数百種類の細胞が適切に混ざり合って構成されています。体の臓器が数十年にわたって正常に働くためには、必要な細胞を必要なだけ供給する幹細胞が必要ですが、臓器には幹細胞がごくわずかしか含まれていません。多種多様な細胞集団や希少な細胞の機能を理解するためには、一つ一つの細胞の特徴を調べる必要があります。その方法として、1細胞ごとにRNAの種類と量を計測する「1細胞RNAシーケンス法(1細胞RNA-seq)[2]」があります。たくさんの細胞で正確に1細胞RNAシーケンスを実施できれば、細胞の状態を正確に計測できます。これまで、大量の1細胞からRNAの種類と量を計測する高出力型1細胞シーケンス法が開発されてきましたが、非高出力型の従来法と比べて、50~60%程度の遺伝子しか捉えられず、希少な細胞の状態を類推することが困難でした。
今回、共同研究チームは、高い検出遺伝子数と低コストを両立した、高出力型1細胞シーケンス法「Quartz-Seq2」を開発しました。Quartz-Seq2は、市販の高出力型1細胞RNAシーケンス法とほぼ同等のコストながら、200~240%の遺伝子数を検出しました。またQuartz-Seq2を利用し、胚性幹細胞(ES細胞)[3]・分化細胞の計4,500個において、数個の希少細胞を検出することに成功しました。さらに、間葉系幹細胞[4]が含まれる約1,000個のマウス脂肪組織から取り出した細胞を1細胞RNAシーケンスしたところ、間葉系幹細胞には2種類の幹細胞が含まれることが分かり、それぞれの細胞機能の違いを類推することができました。
本成果は、細胞分化や臓器・器官発生などの基礎研究から、再生医療における移植細胞の有効性・安全性評価など、様々なライフサイエンスの研究分野の発展に貢献すると期待できます。
本研究は、英国の科学雑誌『Genome Biology』(3月9日付)に掲載されました。
本研究は、科学技術振興機構(JST)および日本医療研究開発機構(AMED)再生医療実現拠点ネットワークプログラム「iPS・分化細胞集団の不均質性を1細胞・全遺伝子解像度で高速に測定する技術の開発」、文部科学省科学研究費補助金、日本学術振興会(JSPS)科学研究費補助金の支援を受けて行われました。また、本研究の一部は、JST戦略的創造研究推進事業(CREST)「臓器・組織内未知細胞の命運・機能の1細胞オミクス同時計測」の支援を受けて行われました。
※共同研究チーム
- 理化学研究所 情報基盤センター バイオインフォマティクス研究開発ユニット
- 上級センター研究員 笹川 洋平(ささがわ ようへい)
- センター研究員(研究当時) 團野 宏樹(だんの ひろき)
- テクニカルスタッフⅠ(研究当時) 海老澤 昌史(えびさわ まさし)
- テクニカルスタッフⅠ 田中 かおり(たなか かおり)
- センター研究員 林 哲太郎(はやし てつたろう)
- ユニットリーダー 二階堂 愛(にかいどう いとし)
(多細胞システム形成研究センター 一細胞オミックス研究ユニット ユニットリーダー) - 奈良先端技術大学大学院 バイオサイエンス研究科 幹細胞工学
- 助教 高田 仁実(たかだ ひとみ)
- 教授 栗崎 晃(くりさき あきら)
背景
私たちの体は、数百種類の細胞が適切に混ざり合って、構成されています。体の臓器が数十年にもわたって正常に働くのは、臓器に含まれる少数の幹細胞が、常に細胞を供給し続けるからです。また、臓器には多種多様な細胞種が含まれており、これらの細胞が互いに関連して臓器の機能を支えています。しかし、これらの幹細胞の発見や、複数細胞の機能的相互作用の理解は十分には進んでいません。
細胞が持つ多様な機能は、ゲノムDNAにコードされた数万種類のRNAの組み合わせによって決まります。RNAは、さまざまなタンパク質に翻訳され、細胞のさまざまな機能を担います。臓器を構成する細胞種を判別し、その機能を類推するには、1細胞が持つRNAの量と種類を計測する必要があります。
これを実現する技術が、「1細胞RNAシーケンス法(1細胞RNA-seq)」です。1細胞RNA-seqでは、RNAをDNAに変換してDNAシーケンサーによって配列決定し、RNAの種類や量を計測します。ただし、一つの細胞にはRNAが10ピコグラム(pg、1pgは1兆分の1グラム)というごくわずかな量しか含まれていません。そこで、微量RNAをDNAシーケンサーで読めるcDNA[5]に変換する「逆転写反応 [6] 」と、cDNAを計測できる量まで分子を増幅する「全cDNA増幅法 [7] 」を実施することが必要になります。
臓器内の多種多様な細胞集団や希少な幹細胞を漏れなく捉えるには、1度の実験で大量の1細胞RNA-seqを実施する必要があります。これを実現するのが、「高出力型1細胞RNA-seq法」です(図1)。高出力型1細胞RNA-seq法は、マイクロ流体装置を利用し、大量の1細胞RNA-seqを実施できます。しかし、これまで開発された実験手法は感度が悪く、非高出力型の従来法と比べて、50~60%程度の遺伝子しか捉えられませんでした。そのため、細胞種類の同定は可能ですが、細胞の詳しい機能を調べることは困難でした。また、マイクロ流体装置の流路を利用し、細胞を観察せずに1細胞をランダムに捕捉するため、その細胞の形状や機能的特徴が分からないまま、シーケンスしなければなりませんでした。
笹川上級センター研究員らは、2013年に検出遺伝子が最も多い高精度の1細胞RNA-seq法「Quartz-Seq」を開発し注1)、世界中で利用されています。しかし、Quartz-Seqは数十個~百個程度の1細胞をそれぞれシーケンスするもので、大量の1細胞を計測できませんでした。そこで、検出遺伝子数が多く、かつ大量の1細胞由来のRNAを計測できる手法の開発が求められていました。

図1 臓器の成り立ちや疾患の理解に貢献する高出力型1細胞RNA-seq法の概念図
ヒトの体は約数十兆個の細胞からなり、数百種類の細胞で構成されているが、臓器の維持に関わる幹細胞は臓器にわずかしかない。このような多様・希少な細胞の機能や状態を調べれば、臓器の成り立ちや疾患を理解できる。しかし、1細胞単位ではなく、臓器単位の実験では、希少な細胞の情報が薄まってしまい、細胞の詳しい機能までは分からない。また、多様な細胞種が混合しているため、細胞種ごとの情報も得られない。そのため、高出力型1細胞RNA-seq法を利用し、臓器を構成する一つ一つの細胞を、大量かつ高速で正確に計測する必要がある。従来法では、たくさんの細胞を実験できるが、検出できる遺伝子数に限界がある。そのため、細胞種類の同定は得意だが、細胞周期などの細胞状態の違いや細胞の持つ様々な機能の同定が不得手だった。Quartz-Seq2は検出できる遺伝子数が多いため、細胞状態や機能の正確な同定が容易になった。
注1)2013年7月25日プレスリリース「細胞1個の遺伝子発現を網羅的に定量化する「Quartz-Seq法」を開発」
研究手法と成果
共同研究チームは、数千~数万の1細胞由来RNAを低コストに計測できる高出力型1細胞RNAシーケンス法「Quartz-Seq2」の開発に取り組みました(図2)。
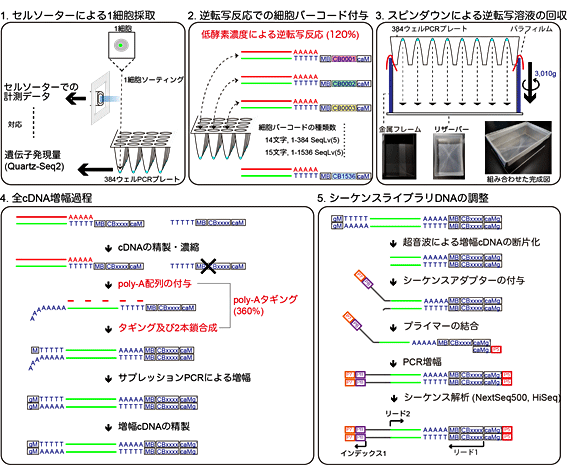
図2 Quartz-Seq2の概要
Quartz-Seq2は、主に五つの工程からなる。
- セルソーターで細胞の情報を取得しつつ、384ウェルプレートに1細胞を採取する。384ウェルプレートの各ウェルには、互いに異なる細胞バーコード配列がついた逆転写プライマーを入れておく。
- 逆転写反応を行い、各ウェルに入った1細胞由来のpoly-A RNAから、細胞バーコード配列の付いたcDNAを合成する。
- 各ウェルのcDNAが入った溶液を、スピンダウン(遠心分離)しリザーバーに集める。ウェルと1細胞の対応は、最終的にシーケンス解析時に、固有の細胞バーコード配列に振り分けることで情報科学的に判別が可能である。
- 一つに集めたcDNAは、精製・濃縮して1チューブで反応が可能である。cDNAはpoly-Aタギングと呼ばれる工程で、増幅可能なcDNAに変換する。これは、まずcDNAの3’側にpoly-A配列を付与し、その配列をのりしろに2本鎖を合成する。その後、必要なDNA量を得るためにPCR増幅をする。
- 得られたcDNAに、シーケンスに必要なアダプター配列を付与する。シーケンスでは、ウェルを認識する細胞バーコード配列とRNA分子をカウントするのに必要な分子バーコードをリード1で読み取り、RNAトランスクリプト配列をリード2で読み取る。
Quartz-Seq2の開発は、2013年に笹川上級センター研究員らが開発したQuartz-Seqを改善することにより実現しました。改善点は、次の三点です。
1)逆転写反応と全cDNA増幅法の効率上昇
一つ目の改善として、逆転写反応と全cDNA増幅法の効率上昇に取り組みました。大量の1細胞のRNAをシーケンスするには、DNAシーケンサーで大量の細胞を計測しなければなりません。しかし、コストに限界があるため、1細胞に割り当てられるシーケンス数が減ります。通常のシーケンス実験では、数十サンプルに対して、それぞれ数千万のDNA配列断片を計測します。しかし、1細胞RNA-seqでは数千個の1細胞をシーケンスするため、1細胞あたり10万程度のDNA配列、つまり通常の1/100程度しか得られません(図3a)。共同研究チームは、少ないシーケンス量(リード数)を最大限に活用するには、細胞内のRNAを漏れなくシーケンスできる分子に変換することが重要だと考えました。
1細胞RNA-seqは、1細胞由来のごく微量なRNAをcDNAに変換する「逆転写反応」と、cDNAを計測できる量まで増幅する「全cDNA増幅法」の二つから構成されています。まず、逆転写効率を上昇させるために、温度条件や反応液を改善し、約20%の効率上昇を達成しました。
次に、全cDNA増幅法に関してQuartz-Seq2では、全cDNAにPCR増幅[8]用の共通配列を付加するステップがあり、poly-Aタギング[9]という反応を用います。さまざまな反応液の組成を検討し、反応効率を上昇させる新しい組成を突き止め、反応温度を段階的に上昇させる装置を利用して、反応効率を上昇させました。これらの改善で、従来のpoly-Aタギングと比べて、約360%も多いcDNAが得られました(図4a)。
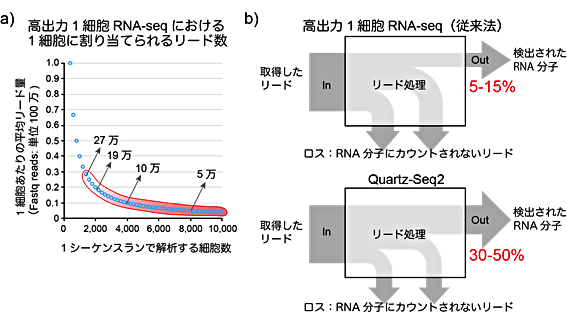
図3 高出力型1細胞RNA-seqで求められる高効率のリード活用
a) 高出力型1細胞RNA-seqは、1回のシーケンス解析で数千の1細胞を解析するため、1細胞あたりに割り当てられるシーケンス量(リード数)が、通常のRNA-seqの1/100以下の10万程度である。
b) 1細胞RNA-seq法では、取得したリードを処理することで、RNA分子の種類と数を検出できる。一方で、従来の高出力1細胞RNA-seq法では、検出RNA分子への変換効率が低い。Quartz-Seq2は、この変換効率を大幅に向上させた。
2)逆転写酵素量の削減によるコスト削減
二つめの改善は、RNAをDNAに変換する逆転写酵素量の削減です。逆転写反応は細胞バーコードを付加するステップなので、必ず1細胞ごとに実施する必要があります。そのため、逆転写酵素はもともと高価な試薬である上に、細胞数分の酵素量を消費することからコストがかかります。また一般的には、酵素反応は、酵素と基質が分子的に混み合う状況の方が反応効率がよいと考えられています。しかし、高価な酵素量を増やすのは難しく、逆に高濃度にするために反応液量を減らすには、特殊な分注装置や容器が必要になります。
そこで、既存の逆転写酵素量から割合を変化させて反応を調べたところ、1.56%~6.25%でも反応が正常に進むことが分かりました(図4b)。これで、反応コストの大部分を占めていた逆転写酵素量を削減できました。また、逆転写反応の効率は分子の混み合いと関連性が薄いと考えられました。
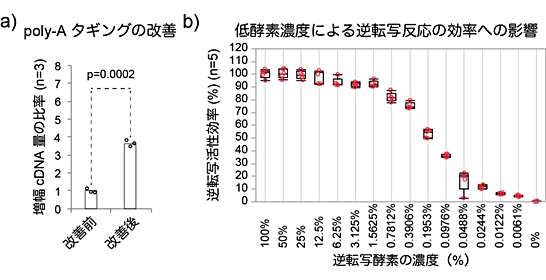
図4 Quartz-Seq2反応の改善点
a)Quartz-Seq2では、poly-Aタギングでの緩衝液や反応条件を最適化することにより、従来と比べて約360%の量のcDNAが得られた。
b)Quartz-Seq2では、逆転写反応の緩衝液や反応条件を最適化し、逆転写効率を約20%向上させた。同条件では、通常の1.56%程度の酵素量であっても、90%以上の逆転写活性の効率があることが分かった。安定して酵素を分取できるのは6.25%以上であるため、同条件をQuartz-Seq2に採用している。
3)DNAバーコード法による実験コスト削減と精度向上
三つめの改善は、DNAバーコード法の利用です。Quartz-Seq2では、細胞バーコード[10]と分子バーコード[11]という二つのDNA標識技術を使用しました。
1細胞ごとに区別して計測するには、1細胞ごとにシーケンスできる分子に変換する必要があります。そのため細胞の数だけ容器や試薬が必要になり、実験コストが高くなります。そこで、RNAをDNAに変換する逆転写反応の際に、1細胞ごとに異なるDNA配列(細胞バーコード)を付加しました。細胞バーコードを付加した後に、全ての細胞由来のDNAを混合し反応させて、シーケンスを行っても、バーコードを頼りにコンピュータ上で、1細胞ごとのデータに分離できます。Quartz-Seq2では、384または1,536種類の細胞バーコードを利用し、これらの1細胞由来cDNAを混合して、全cDNA増幅反応ができるようにしました。これにより、容器や試薬量の削減に成功し、細胞バーコードを用いない市販品の方法に比べて、実験コストを約1/100に削減できました。
また、これらの細胞バーコードは、情報科学的に誤りを補正できるよう設計されているため、シーケンス決定配列やDNA合成時の配列の置換だけでなく、従来法では補正できなかった挿入・欠失も補正できます。正確なDNAバーコードの合成には約1,400万円の費用がかかりますが、今回の改善により、合成エラーが含まれる安価な合成法を利用できることから、約40万円で細胞バーコードの合成が可能になりました。
分子バーコードは、RNA分子一つ一つを区別できるバーコードです。1細胞RNA-seqでは、さまざまな長さや配列を持つRNA分子をcDNAに逆転写した後、計測できる量までPCRで全cDNA増幅します。この際に、増えやすいRNAと増えにくいRNAが存在するため、増幅率は遺伝子によって偏ります。ただし、いくら偏りが生じても、分子バーコード数を数えれば、偏りの影響を受けずに分子数を特定できます。分子数計測の実験エラーは、理論的にはポアソン分布[12]に従います。Quartz-Seq2は、分子バーコードを利用して実験エラーを除くと、ほぼポアソン分布に従うことが分かりました(図5a)。このことは、そもそも増幅時の実験エラーが非常に少ないことを示しています。十分な量のRNAを利用した、通常のRNA-seqと比較しても高い相関を示しました(図5b)。
これら二つのバーコード技術により、数千個の1細胞から数千の遺伝子を正確に捉えることができ、実験も3日程度という短期間で終えられました。
以上の改善により、Quartz-Seq2は、市販の高出力型1細胞RNA-seq法とほぼ同等のコストながら、200~240%多くの遺伝子数を検出しました(図5c)。また、従来の非高出力型1細胞RNA-seq法と比較しても、3~28%程度のコストでありながら、検出遺伝子数が約130~170%多いことを示しました。
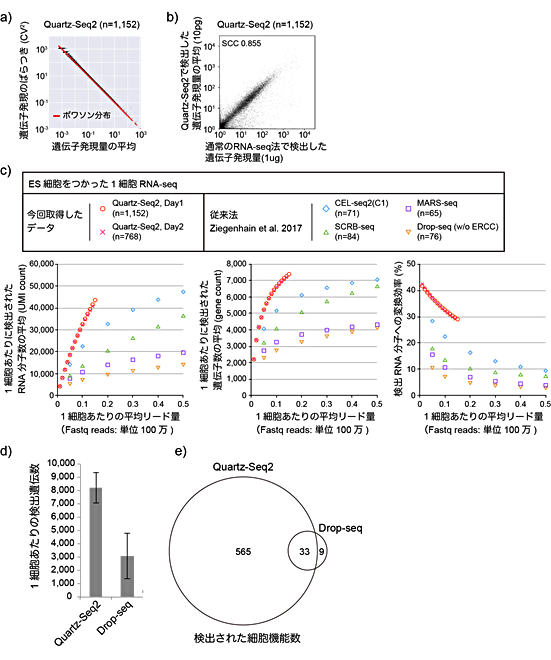
図5 Quartz-Seq2の性能
a)1細胞に相当する10pgのRNAを使用した繰り返し実験(n = 1,152)。黒点は遺伝子、Y軸は1,152ウェル間の遺伝子発現量のばらつき、X軸は遺伝子発現量の平均を示す。Quartz-Seq2での遺伝子発現のばらつきは、ほとんどポワソン分布(赤線)と重なり、繰り返し実験間のエラーが極めて少ないことを示している。
b)Quartz-Seq2と1マイクログラム(μg、1μgは100万分の1グラム)のRNAを使った通常のRNA-seq法の比較。高い相関を示した。
c)ES細胞を使った従来法とQuartz-Seq2の性能比較。Quartz-Seq2(赤丸)では、従来法よりも検出RNA分子数や検出遺伝子数が多い。シーケンス量を増やしても、従来法がQuartz-Seq2の性能に追い付くのは困難。
d)ES由来細胞を使ったDrop-seqとQuartz-Seq2の比較解析。Drop-seqは既存の高出力型1細胞RNA-seq法。データは1細胞あたり24万リードを割り当てた場合、それぞれの方法で検出される1細胞あたりの遺伝子数。Quartz-Seq2がDrop-seqよりも約260%多い。
e)高変動遺伝子が担う細胞機能の数の重なり。Quartz-Seq2は、Drop-seqより多くの細胞機能を検出できる。
共同研究チームは、検出遺伝子数が増えることで、細胞の中に含まれるさまざまな機能を観測できることを示しました。細胞は増殖や分化、代謝などさまざまな機能の組み合わせで構成されています。それぞれの機能は複数の遺伝子の組み合わせで実現されます。つまり、検出できる遺伝子が多いと、検出する機能も多くなると考えられます。
そこで、ES細胞と、ES細胞から分化させた細胞種をそれぞれ2,000細胞ずつシーケンスしました。得られた結果から、それぞれの細胞を特徴づける遺伝子の数を調べたところ、従来法と比べて約260%多く得られました(図5d)。ところが、検出されたそれぞれの遺伝子が担う機能数は、14.7倍も多いという結果になりました(図5e)。細胞の状態や発揮する機能を明らかにするには、検出遺伝子数と低コスト、高出力を兼ね備える必要があると考えられます。
また、分子バーコード変換効率という指標を用いると、1細胞RNA-seq法の性能を評価できることも示されました。これは、得られたシーケンスデータがどれだけ有効に利用されたかという指標です。1細胞RNA-seq法では、得られたシーケンスデータのうち、分子バーコードの重複や増幅時に生じた副産物などにより、シーケンスデータの一部が有効に利用できません。特に高出力型1細胞RNA-seq法では、少ないシーケンス量を1細胞ごとに分け合うため、シーケンスデータを無駄にできません。本研究では、従来法では5~15%のところ、Quartz-Seq2では30~50%であることを示しました(図3b)。また、この指標はシーケンス量や細胞あたりのRNAに強く依存するため、これらの条件を揃えて比較することが重要と考えられます。
次に、Quartz-Seq2の応用例として、細胞集団からの希少細胞の検出や細胞の状態の検出が可能であるか実験しました。まず、ES細胞とその分化細胞、計約4,500細胞分のシーケンスをしたところ、8個の希少細胞(Zscan4c/d)を捉えることに成功しました(図6a)。この結果は、再生医療で用いられる移植用の細胞に混入してしまう目的外細胞の有無の検査などに利用できることが期待されます。また、Quartz-Seq2は1細胞をセルソーター[13]で採取するため、細胞の形状やDNA量などの情報が利用できます。また、細胞周期に伴って変動するRNAとDNA量の増減の関係を調べました。細胞周期に伴い変動するRNAは2倍程度の変動しかないため、高出力型1細胞RNA-seq法で捉えるのは困難でしたが、Quartz-Seq2では正確に捉えました(図6b)。セルソーターで得られたDNA量を頼りに細胞周期の順に細胞を並べた後、細胞周期の関連遺伝子のRNAが周期的に発現している様子を観測できました。
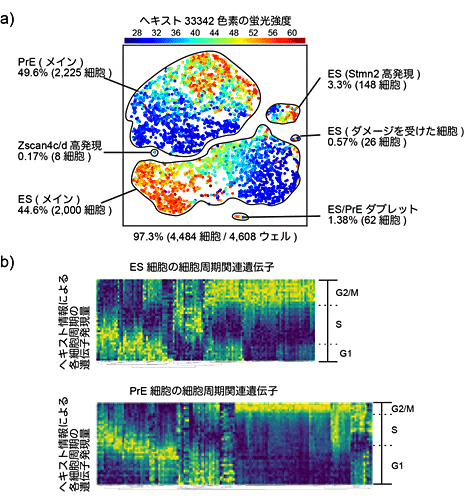
図6 Quartz-Seq2による細胞集団に含まれる希少細胞の検出例
a)Quartz-Seq2による、ES細胞とES細胞由来分化細胞の解析例。培養の際に、0.17%とごくわずかに含まれる細胞集団(Zscan4c/d高発現)の検出に成功した。色は、セルソーターで検出したヘキスト染色の蛍光強度。細胞が持つDNA量を示しており、細胞周期を反映する。
b)ヘキスト染色の蛍光強度を細胞周期の指標として使い、細胞周期で関連遺伝子のRNAが周期的に発現している様子が観測できた。
続いて、間葉系幹細胞が含まれる約1,000個のマウス脂肪組織から取り出した細胞をシーケンスしました(図7)。この細胞群には、複数種類の免疫細胞や間葉系幹細胞が含まれていると言われています。そのうち間葉系幹細胞は、細胞治療の材料として注目されていますが、実際に、どのような細胞種がどの程度含まれているか理解が進んでいませんでした。本研究では、間葉系幹細胞には2種類の幹細胞が含まれることを示し、それぞれの細胞型を選り分ける遺伝子を発見し、細胞の機能的な違いを類推できることを示しました(図7c)。また、今回実施した二つの実験により、Quartz-Seq2が希少細胞を検出し、複数種類からなる臓器の細胞機能を計測できることも示しました。
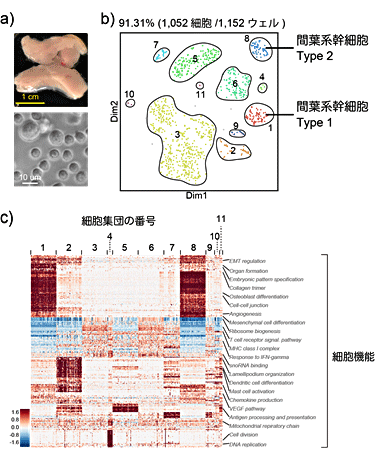
図7 複数の細胞からなる臓器由来細胞を用いたQuartz-Seq2の解析例
a)脂肪組織(上)から間質血管細胞群(Stromal vascular fraction, SVF)という細胞集団(下)を採取。SVFは、細胞径が大きく異なる細胞から成り立っている(下)。
b)Quartz-Seq2によるSVFの1細胞解析結果。約1000個の細胞を解析した結果、11種類の細胞集団を発見した。遺伝子発現のパターンから、2種類の間葉系幹細胞(細胞集団1と8)が存在することが分かった。
c)発見した11種類の細胞集団の中に特異的に利用されている遺伝子があることが分かった。それらの遺伝子が多用な細胞機能に関連することを見出した。
今後の期待
近年、iPS細胞由来分化細胞や体性幹細胞の移植を目指す再生医療が急速に発展しています。しかし、培養状態や分化方法などにより、細胞集団に亜集団が生じます。このような細胞集団の不均一性の計測が可能になれば、安全で有効な再生医療の実現を加速することが期待されます。大量の1細胞由来RNAを網羅的、高精度かつ低コストで計測し、1細胞の特徴を捉えられるQuartz-Seq2は、細胞集団に含まれる細胞亜集団やその遺伝子発現マーカーの発見に貢献するものです。また、がんは少数の細胞に徐々に変異が蓄積し、細胞集団が不均一になります。がん細胞集団の不均一性をQuartz-Seq2の技術で計測することで、早期に変異した細胞を発見し、それを標的とする創薬が行えるようになると期待できます。
今回、共同研究チームは、Quartz-Seq2を開発する過程で、酵素量削減やポリA付加反応の向上、DNA回収装置なども開発しました。これらの反応や装置は汎用的であり、従来法の1細胞RNA-seq法にも応用できるため、1細胞RNA-seq法を開発・実施する研究へ貢献すると期待できます。
論文情報
- タイトル
- Quartz-Seq2: a high-throughput single-cell RNA-sequencing method that effectively uses limited sequence reads
- 著者名
- Yohei Sasagawa, Hiroki Danno, Hitomi Takada, Masashi Ebisawa, Kaori Tanaka, Tetsutaro Hayashi, Akira Kurisaki, and Itoshi Nikaido
- 雑誌
- Genome Biology
- DOI
- 10.1186/s13059-018-1407-3
補足説明
- [1] 高出力型1細胞RNAシーケンス法「Quartz-Seq2」
- 大量の1細胞由来のRNAをシーケンスできる技術を高出力型1細胞RNAシーケンスと呼ぶ。Quartz-Seq2(クォーツ・セックツー) は、数千から数万個の1細胞由来のRNAをシーケンスすることで、細胞機能や特徴を明らかにできる計測手法。本共同研究チームの笹川と二階堂が2013年に発表したQuartz-Seqを基盤に、たくさんの細胞由来の1細胞RNAをシーケンスできる方法に改善された。
- [2] 1細胞RNAシーケンス法(1細胞RNA-seq)
- 1細胞中に含まれるRNAをDNAシーケンサーを用いて配列決定し、網羅的かつ定量的にその量や種類を決定する方法。微量なRNAを用いるため、微量RNAからcDNAを合成する「逆転写反応」と、シーケンス可能な量までcDNAを増幅させる「全cDNA増幅法」の二つのステップからなる。
- [3] 胚性幹細胞(ES細胞)
- 哺乳類の着床前胚(胚盤胞)に存在する内部細胞塊から作製した細胞株で、体を構成する全ての種類の細胞に分化する能力(多能性)を持つもの。
- [4] 間葉系幹細胞
- 成体幹細胞の一つで、脂肪や骨髄、臍帯、滑膜など中胚葉性組織(間葉)に存在する。骨、軟骨、神経、脂肪などに分化する能力があり、再生医療への応用が期待されている。
- [5] cDNA
- RNAを鋳型に逆転写酵素で合成されたDNA。RNAに対して相補鎖になるため、相補的DNA(complementary DNA)と呼ばれる。
- [6] 逆転写反応
- RNAを鋳型とし相補的なDNA(cDNA)を合成する反応。RNAウイルスが宿主細胞に進入し自己複製する際に働く、RNA依存性DNA合成酵素である逆転写酵素を利用する。逆転写酵素がcDNA合成を行うには、RNA配列に相補的に結合した短いDNA鎖(逆転写プライマー)を出発点とする必要がある。現在、よく使われているシーケンサーは、DNAしか読めないので、RNAを計測するにはDNAに変換する必要があり、逆転写反応が利用される。
- [7] 全cDNA増幅法
- 全てのRNA分子を増幅する技術。RNAを増幅できる分子に変換するステップと、増幅するステップに分けられる。前者は、RNAから1本鎖cDNAに逆転写するときに、増幅用の配列を付加するステップと、1本鎖cDNAを2本鎖cDNAに変換するステップで、増幅用配列を付加する反応からなる。Quartz-Seq2では2本鎖cDNA合成時にpoly-Aタギングという方法を利用している。その他にテンプレートスイッチング法などがある。増幅のステップでは、前のステップで2本鎖cDNA両端に付加された増幅用配列を利用してPCR増幅を行う。
- [8] ポリメラーゼ連鎖反応(PCR)
- 熱耐性を持つDNA依存性DNA合成酵素を用いて、鋳型DNAとPCRプライマーの結合、相補DNAの合成、二本鎖DNAの解離の3ステップを繰り返すことで、DNAを連続的かつ指数関数的に増幅させる反応。鋳型DNAの長さや配列に依存した増幅バイアスが生じる。長いDNAは増えにくい。さまざまな種類のRNAを由来とするcDNAを増幅させるには、PCRプライマーが結合するための共通配列をあらかじめ逆転写プライマーに付加させておく必要がある。PCRはpolymerase chain reactionの略。
- [9] poly-Aタギング
- ターミナルトランスフェラーゼという酵素によって、DNAの末端に複数個のアデニン(poly-A)が付加される反応を利用して、DNA末端にタグを付加する反応。DNA末端に複数のアデニンを付加することで、相補な複数のチミン(poly-T)を用いて、相補鎖の合成の起点とできる。全cDNA増幅では、逆転写したcDNAの3’末端をpoly-Aを起点に2本鎖cDNA合成をしつつ、PCR用のプライマー配列(タグ)を付加する。
- [10] 細胞バーコード
- 1細胞ごとに異なる数文字から十数文字のDNA配列。逆転写時にこの配列を付加しておけば、その後は混合して反応、DNAシーケンスができる。得られたDNA配列に含まれる細胞バーコードを頼りに、シーケンスリードを分類することで、元の1細胞ごとのデータを復元できる。スウェーデンのIslamらが1細胞RNA-seqに導入した技術。
- [11] 分子バーコード
- RNA分子ごとに異なる8~10文字のDNA配列。逆転写プライマーにランダムな配列を入れておくことで、RNA1分子につき、1種類のバーコードが付加される。RNA分子あたり一つのバーコードしか出てこないため、もしバーコードが重複していたら、PCRの増幅の偏りだと考えられる。そのため、バーコードの重複を除けば、RNA分子数をカウントできる。2011年、フィンランドとスウェーデンの研究チームのKiviojaらが提案。
- [12] ポアソン分布
- ある時間中に平均でλ回発生する事象が、k回発生する確率を表わす分布。ある一定時間中に事故が起きる確率などを表わすときなどに使われる。RNA分子からシーケンスを観測できる確率もポアソン分布で表わすことができる。
- [13] セルソーター
- 細胞を分取する装置。液滴に細胞を閉じ込めて落下させ、レーザーで励起することで、その細胞の特徴を計測する。計測データに基づき、目標とする1細胞を1容器に分取できる。機種によっては、384や1536ウェルの容器に1細胞を採取することも可能。
発表者・機関窓口
発表者
理化学研究所 情報基盤センター バイオインフォマティクス研究開発ユニット
上級センター研究員 笹川 洋平(ささがわ ようへい)
センター研究員(研究当時) 團野 宏樹(だんの ひろき)
ユニットリーダー 二階堂 愛(にかいどう いとし)
機関窓口
理化学研究所 広報室 報道担当
AMED事業に関するお問い合わせ先
日本医療研究開発機構(AMED)
戦略推進部 再生医療研究課
