2022-04-26 国立精神・神経医療研究センター
国立精神・神経医療研究センター(NCNP)神経研究所免疫研究部の大木伸司室長、山村隆特任研究部長、東京工業大学生命理工学院の林宣宏教授、同大学院生高橋文緒らの研究グループは、神経変性疾患に共通した病態伝播・拡散のしくみとして、免疫細胞が引き起こす新しい神経細胞障害メカニズムを明らかにし、これまでタンパク質の異常凝集による神経細胞死を中心に展開してきた神経変性疾患研究に、新しい病態制御の可能性を示す研究成果を発表しました。
研究成果は、「iScience(アイ・サイエンス)」オンライン版に2022年4月23日午前0時(日本時間)に掲載されました。
研究の背景
神経変性疾患は、筋萎縮性側索硬化症(ALS)、アルツハイマー型認知症(AD)などを含む、中枢神経系の難病です。例えばALSでは、脳脊髄に分布する運動ニューロンが選択的に障害されることで、運動機能の異常を呈し、ADでは、記憶を司る海馬周囲の神経細胞の障害により、認知機能が損なわれます。臨床症状や経過は疾患ごとにさまざまですが、ALSではTDP-43、SOD1、FUSなどのタンパク質が、ADではアミロイド(A)やTauなどのタンパク質が、それぞれ神経細胞内外で凝集体を形成して神経細胞死を引き起こすことが、主要な原因と考えられています。ところが最近、細胞外タンパク質凝集体であるAの除去が、必ずしもAD患者の症状改善につながらないことや、凝集タンパク質が形成される位置と病変部位が必ずしも相関しないことなどがわかってきました。さらに遺伝などの影響なしに発症する患者では、検査などを通じて凝集タンパク質の種類を特定することも困難はあり、治療戦略を考える上で大きな障壁となっていました。最近では、脳内の神経細胞以外の細胞(グリア細胞など)の異常な挙動が、各神経変性疾患に共通して神経細胞死に関わるという考え方が広がっています。神経変性疾患ごとに独立した原因を探ることで治療法に結びつけることを目指す研究だけでなく、種々の神経変性疾患に共通したイベントから、これまで見過ごされてきた原因を明らかにするような研究にも目が向けられるようになってきました。その様な状況下で私たちは、以前に発見した免疫細胞の機能異常が引き起こす神経細胞死のメカニズムが、複数の神経変性疾患でも共通に認められる神経細胞障害のメカニズムであるかどうかを検証するための研究を進めてきました。
研究の概要
多発性硬化症(MS)は、神経軸索を取り巻く髄鞘の障害によって生じる脱髄疾患ですが、MSの亜型である二次進行型MS (SPMS)では、神経変性疾患と似た神経細胞障害の病理像や経過を示すことが知られています。研究グループは、以前にSPMSのモデルマウスを確立し、同モデルの解析を通じてSPMSの中心的な病態である神経細胞障害に、エオメス陽性ヘルパーT細胞という免疫細胞が重要な役割を果たすことを報告しました(Raveney et al. Nature Comm 2015)。エオメスヘルパーT細胞は、グランザイムBというタンパク分解酵素を分泌して、神経細胞死を引き起こしします。研究グループは、SPMS患者の血中エオメス陽性ヘルパーT細胞頻度が、障害進行度とよく相関すること、SPMS患者の脳内に同細胞が広く分布し、大部分がグランザイムBを発現することを示し、神経細胞死を引き起こすSPMSの病原性細胞であることを明らかにしました(Raveney et al. PNAS 2021)。エオメス陽性ヘルパーT細胞は、炎症環境が引き金となって生成することから(Zhang et al. PNAS 2019)、研究グループは、慢性炎症を伴う神経変性疾患にも、同細胞が関わるのではないかと考えました。

図1 mSOD1マウス・5xFADマウスの中枢神経系へのエオメス陽性ヘルパーT細胞の集積
そこで、ALSとADの病態モデルマウスであるmSOD1マウスと5xFADマウスを用いて免疫細胞の脳内集積を調べたところ、発症に伴ってエオメス陽性ヘルパーT細胞が増えていることがわかり、病態との関連が示されました(図1)。さて、エオメス陽性ヘルパーT細胞がグランザイムBを放出するには、抗原を介した活性化が必要と考えられるため、研究グループは、エオメス陽性ヘルパーT細胞を活性化する抗原の探索を行いました。その結果、ゲノム の半分近くにも及ぶトランスポゾンといわれる塩基配列の中で、レトロウイルス様の生活環を持つレトロトランスポゾンの1つであるLINE-1(長鎖散在反復配列-1、Long interspersed nuclear element-1、L1)がコードするORF1 というタンパク質が、エオメス陽性ヘルパーT細胞を活性化することを発見しました。L1は、進化の過程で自身の遺伝子断片をコピー&ペーストの要領で複製する「レトロ転位」を繰り返し、総延長がヒトゲノム全体の20%弱にもおよぶ領域を占め、その中には50万コピーにも及ぶL1 遺伝子断片を含みます(図2)。実は、神経細胞におけるL1の活性化は、神経変性疾患に共通する特徴的な反応として良く知られていましたが、その意義はよくわかっていませんでした。神経細胞のL1活性化により発現したORF1タンパク質が、エオメス陽性ヘルパーT細胞を活性化するのであれば、結果的にグランザイムBが放出されて、神経細胞が死ぬ可能性が考えられます。そこでSPMSモデルマウス、mSOD1マウスと5xFADマウスの3種類のモデルマウスの神経細胞で、L1の活性化によるORF1タンパク質が増えているのか、について調べたところ、いずれのモデルマウスでも、神経細胞のL1活性化によりORF1タンパク質の量が増えていることがわかりました(図3)。興味深いことに、L1活性化のメカニズムはマウスごとに異なっており、SPMSモデルマウスではエピジェネティックな変化と、芳香属炭化水素受容体(Aryl hydrocarbon receptor, AhR)の活性化が関わっていました。一方、mSOD1マウスでは、神経細胞の細胞周期が部分的に進むことで、L1活性化が生じることがわかりました。
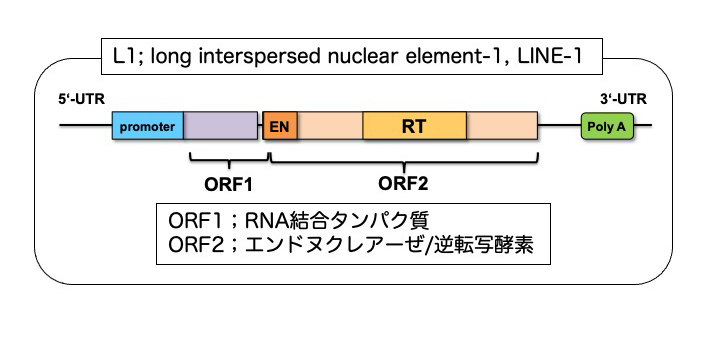
図2 L1レトロトランスポゾンの遺伝子構造
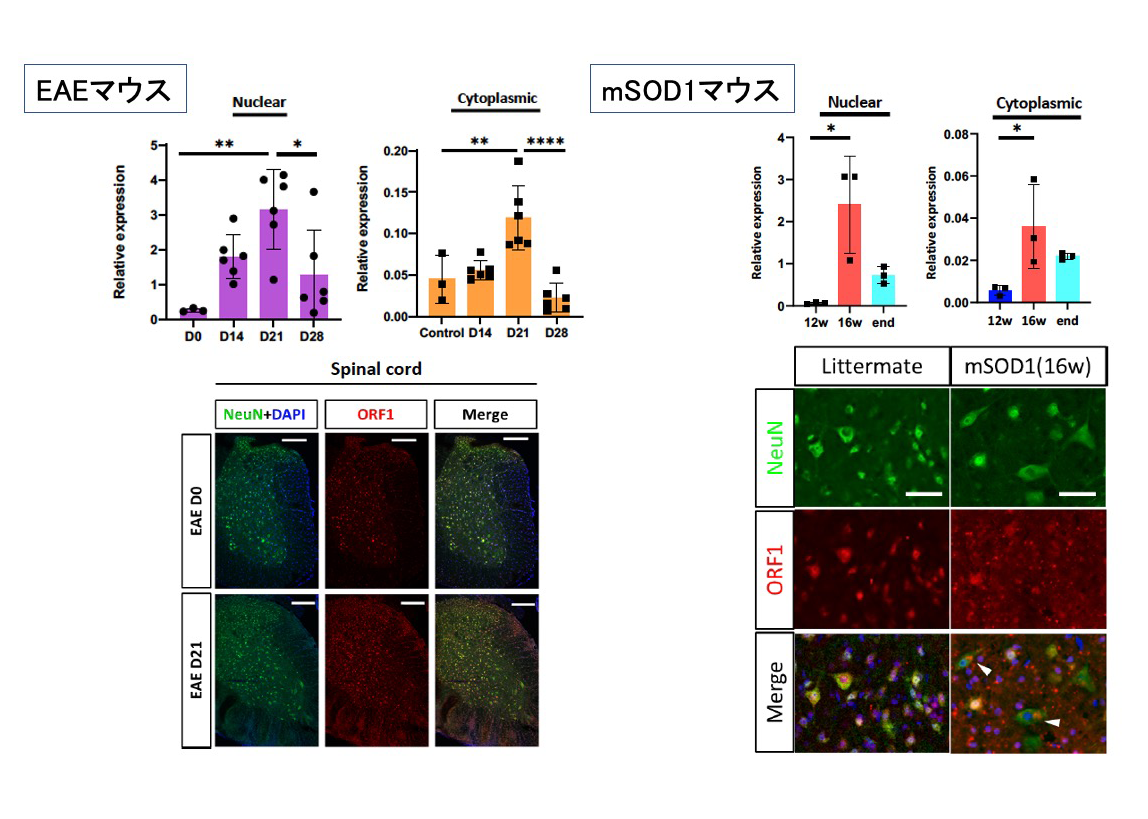
図3 EAEマウスの脊髄神経細胞におけるORF1発現誘導(上段;L1遺伝子発現、;下段ORF1タンパク質発現、いずれも脊髄スライス)
最後に研究グループは、ORF1タンパク質によって活性化したエオメス陽性ヘルパーT細胞が、グランザイムBの放出を介して神経細胞死を引き起こすかどうかを調べました。まずマウスの中枢神経系からTh細胞を分離し、ORF1で刺激すると、グランザイムBの放出が明らかに増えることがわかりました(図4)。さらに分離したTh細胞を、初代培養神経細胞と共にin vitroで培養すると、神経細胞の生存率が明らかに低下しました(図5)。
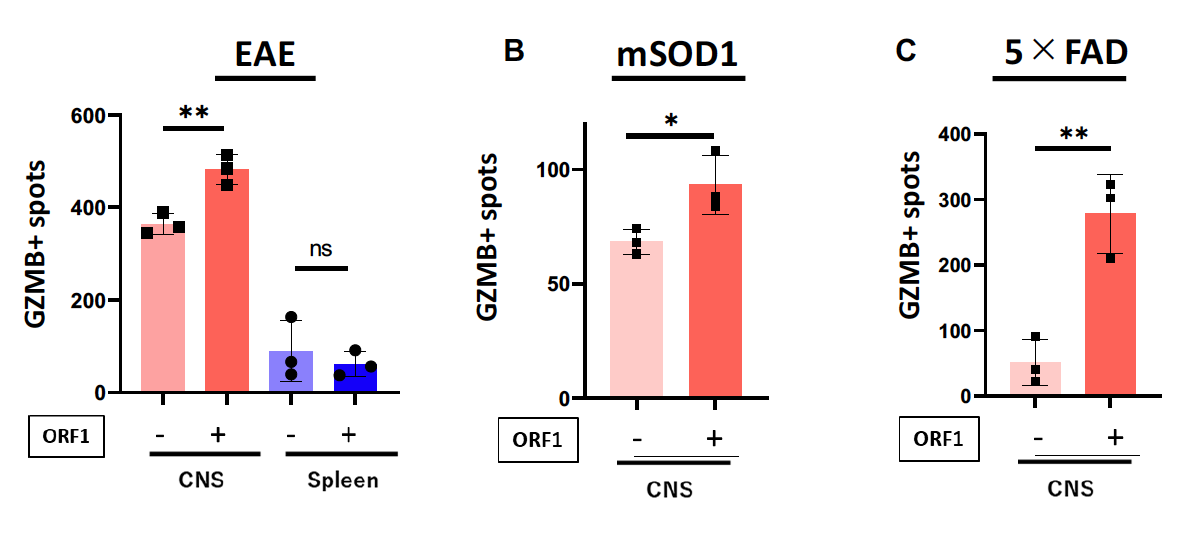
図4 エオメス陽性ヘルパーT細胞のORF1依存的グランザイムB産生
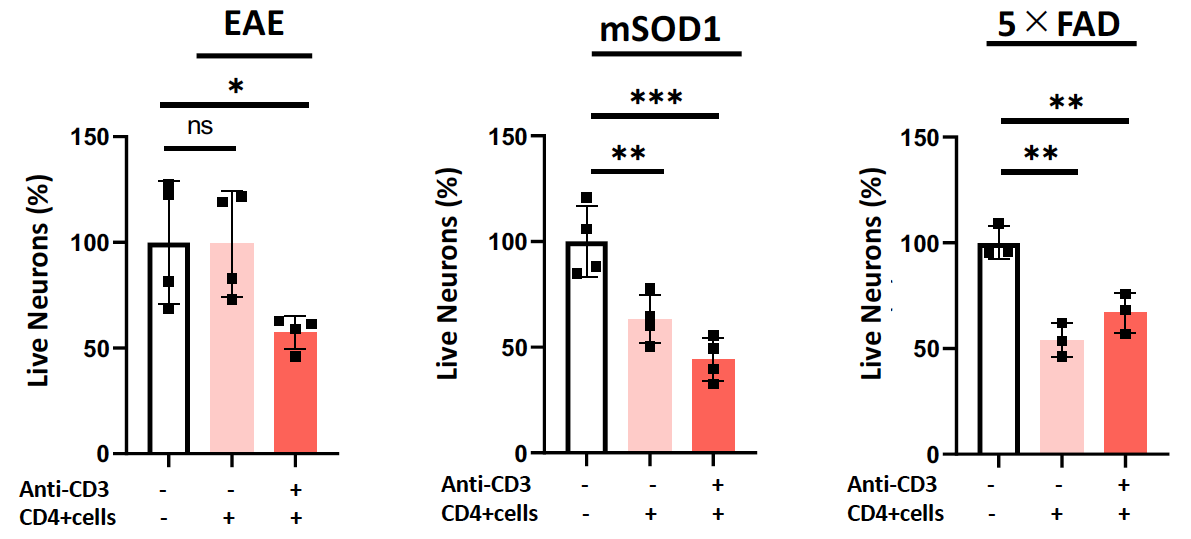
図5 エオメス陽性ヘルパーT細胞による直接的な神経細胞死の誘導
以上の結果から、3種のモデルマウスの中枢神経系において、神経細胞由来のORF1タンパク質がエオメス陽性ヘルパーT細胞の活性化を促し、放出されたグランザイムBが神経細胞死を引き起こすことが示されました。ORF1は元々神経細胞の内部に分布するタンパク質なので、最初に何らかの理由で神経細胞が細胞死を起こすと、細胞外に放出されたORF1がエオメス陽性ヘルパーT細胞を活性化し、周囲に拡散したグランザイムBが、さらに近くの神経細胞にダメージを与える、という反応が繰り返されることで、神経細胞障害が伝播、拡大していくことが明らかとなりました。
本研究により明らかになった免疫依存性神経細胞障害によって、神経細胞死が繰り返されることにより、最初は微小な領域にとどまっていた病変部位が進行性に拡大することで、臨床症状が顕在化して神経変性疾患が発症する可能性が示されました。
今後の展望
今回の研究結果から、免疫細胞が引き起こす神経細胞障害が、神経変性疾患における病変部位の拡大に重要な役割を果たすことがわかりました。これまで神経変性疾患研究においては、凝集タンパク質の蓄積によって生じる神経細胞死の解析が主流でしたが、今回の研究で明らかにした免疫細胞が引き起こす神経細胞障害機序がここに加わることによって、わずかに生じた初期の(凝集タンパク質依存的な)神経細胞死が引き金となって、ORF1抗原の放出とこれを介した免疫細胞が引き起こす神経細胞障害が発動することがわかりました。そしてORF1抗原の放出と免疫細胞が引き起こす神経細胞死が繰り返されることにより、神経細胞死を起こした領域が、周辺へと伝播、拡大していく分子機序が明らかとなりました(図6)。すなわち、神経変性疾患の発症が、初期の(凝集タンパク質依存的な)神経細胞死と、免疫細胞が引き起こす神経細胞死の2つのステップの組み合わせからなると仮定することで、これまで凝集タンパク質の除去だけでは十分な治療効果が得られなかった理由も説明可能となり、神経変性病態の新たなメカニズムが明らかとなりました(図7)。今後は、免疫細胞が引き起こす神経細胞障害を制御する方法の探索、L1の活性化が(凝集タンパク質依存的な)神経細胞死にも関与する可能性を調べることにより、神経変性の伝播、拡大を抑えて、病変部位の拡大による臨床症状の顕在化を回避するための、新しい治療戦略の開発が期待されます。
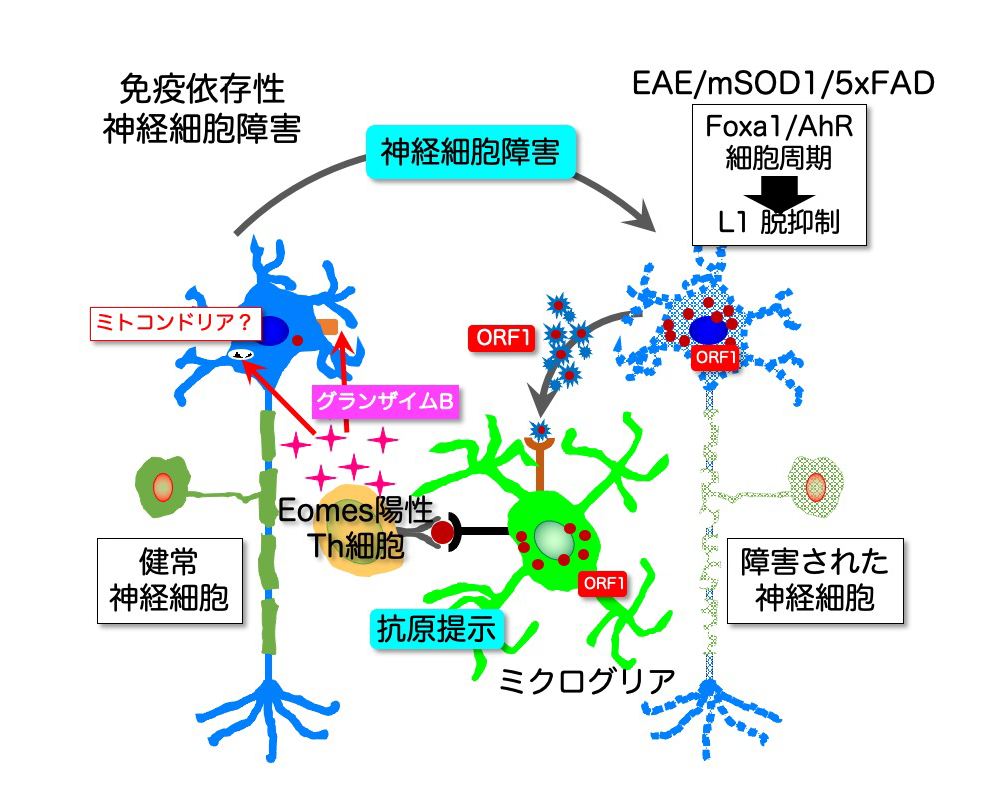
図6 免疫依存性神経細胞障害
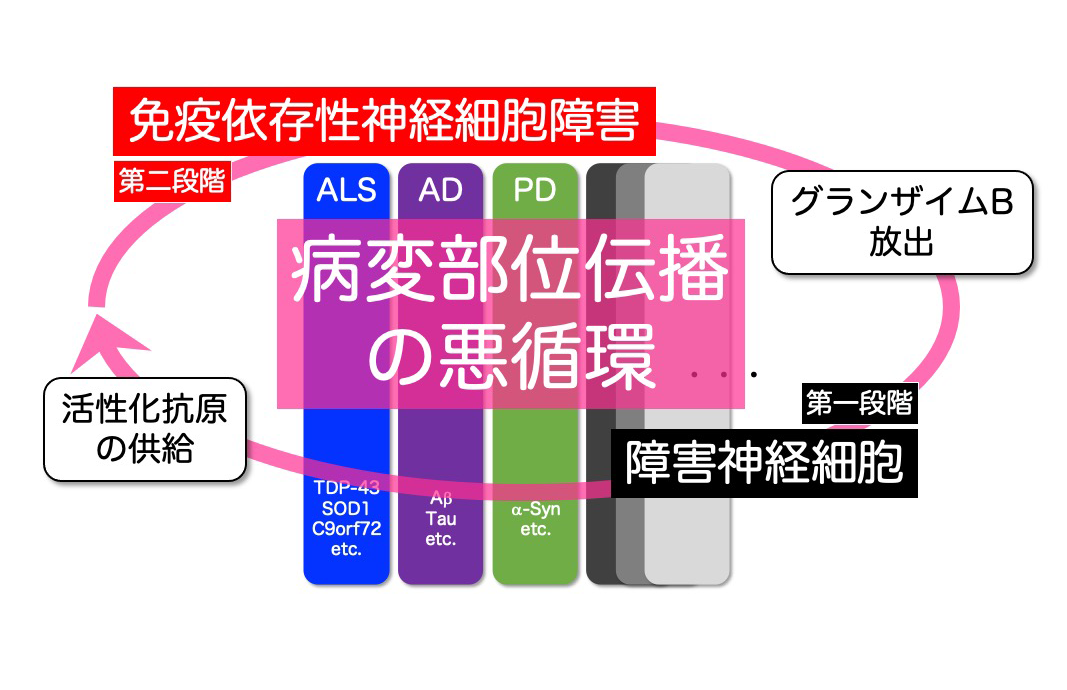
図7 神経変性病態の二段階仮説
前述のとおり、先行する神経細胞死によるORF1分子の供給がなければ、免疫細胞に依存した神経細胞障害は作動しないため、L1の活性化が(凝集タンパク質依存的な)神経細胞死にも関与する可能性についても、今後調べていく必要があると考えています。凝集タンパク質依存的な神経細胞死に対する、これまでの長年にわたる研究成果の蓄積に加えて、今回明らかとなった免疫細胞が引き起こす神経細胞死をコントロールする方法を新しい角度から探索することで、これまで手が届かなかった神経変性疾患の画期的な治療法の開発が期待されます。
用語説明
注1)多発性硬化症(multiple sclerosis; MS)
中枢神経系の炎症性脱髄疾患であるRRMS(再発寛解型MS;relapsing-remitting MS)の他に、RRMSから移行する進行性病態であるSPMS(二次進行型MS;secondary progressive MS)などの病型が知られている。SPMSでは、神経変性疾患様の顕著な神経変性と脳萎縮を生じるが、最近、リンパ球の体内循環を抑制する作用を持つシポニモドがSPMSに有効であることが示され、first-in-classのSPMS治療薬として承認、処方された。SPMSに生じる神経変性による顕著な脳萎縮の機序は長い間不明であったが、シポニモドの有効性が示されたことで、獲得免疫系の関与がクローズアップされつつある。
注2)筋萎縮性側索硬化症(Amyotrophic lateral sclerosis; ALS)
上位および下位運動ニューロンが散発性・進行性に脱落する神経変性疾患である。随意筋の萎縮と運動機能障害が急速に進行し、多くの患者は主に呼吸障害により発症後3〜5年で死亡もしくは侵襲的換気が必要となる。国内の患者は1万人弱とされる。発症者の5~10%を占める家族性ALSでは約20種類の原因遺伝子が同定されているが、発症メカニズムの解明には至っておらず、現時点で有効な治療法が確立していない。患者の90%以上を占める孤発性症例においては、家族性ALSと共通する病態機序が想定されており、近年、孤発性ALS症例の大部分に、TDP-43の機能異常に関連した病理像が見出されたことから、大きな注目を集めている。
注3)アルツハイマー型認知症(Alzheimer’s disease; AD)
認知機能の低下や人格の変化を主な症状とする認知症の一種であり、認知症全体の2/3ほどを占める。ADでは、病変が海馬を中心とした側頭葉内側部から連合野に広がり、この過程で見当識障害、記憶障害、言語障害などの中核症状が出現する。病理増としては、凝集したアミロイドβ(Aβ)からなる老人斑や、リン酸化タウからなる神経原線維変化を多数生じる。家族性ADは全体の1%以下と推定されており、ほとんどが孤発性である。Aβ蓄積を起点とするアミロイドカスケード仮説をはじめ複数の病態仮説が提唱されているが、多因子性疾患と予想されるADの病因の特定は難しく、複合的な要因が重なることで発症にいたると考えられている。
注4)長鎖散在反復配列-1 (Long interspersed nuclear element-1; LINE-1, L1)
自律的な転位能を持つnon-LTR型のレトロトランスポゾンであり,1ゲノム中に50万コピーの遺伝子断片を含み、その総延長はゲノム全体の20%ほどにも及ぶ。完全長のL1は約6kbのサイズを持ち、ORF1とORF2の2つのタンパク質をコードする。ORF1はRNA結合タンパク質であるが,その機能の詳細は不明である。一方、ORF2 はレトロ転位に必要なエンドヌクレアーゼと逆転写酵素の活性を持つ。ヒトでは80-100コピー、マウスでは約300コピーのL1が転位活性を維持している。レトロ転位により機能的遺伝子の発現が阻害されることで、血友病や家族性大腸ポリポーシスなど様々な疾患の原因となることが知られている。
注5)レトロ転位
レトロトランスポゾン (Retrotransposon) は可動遺伝因子の一種であり、自身のRNAから逆転写酵素によって合成した DNAがゲノムに挿入されることで「転位」する。遺伝子近傍または遺伝子内への挿入が起こると、突然変異によって血友病、家族性大腸ポリポーシスやレット症候群など様々な疾患の原因となる。転位活性を維持したゲノム因子であるL1は、脳の発生期間を通じて転位を生じ、神経細胞の遺伝子発現や機能修飾に関わっている。
原著論文情報
・タイトル
Immune-mediated neurodegenerative trait provoked by multimodal derepression of long-interspersed nuclear element-1
・著者名
Fumio Takahashi, Chenyang Zhang, Hirohiko Hohjoh, Ben Raveney, Takashi Yamamura, Nobuhiro Hayashi, Shinji Oki
・雑誌
iScience
・DOI; https://doi.org/10.1016/j.isci.2022.104278
助成金
本研究成果は、以下の事業・研究領域・研究課題によって行われました。
・文部科学省科学研究費補助金
・三菱財団研究助成
・国立精神・神経医療研究センター精神・神経疾患研究開発費
お問い合わせ先
【研究に関する問い合わせ】
国立研究開発法人国立精神・神経医療研究センター
神経研究所 免疫研究部
室長 大木 伸司
【報道に関するお問い合わせ】
国立研究開発法人国立精神・神経医療研究センター
総務課広報係

