

2018/08/28 国立大学法人 弘前大学,京都大学,国立研究開発法人 日本医療研究開発機構
弘前大学小児科学講座の土岐力講師、伊藤悦朗教授らのグループは、京都大学医学部腫瘍生物学講座の吉田健一助教、小川誠司教授らとの共同研究により、新たな先天性骨髄不全症を発見しました。
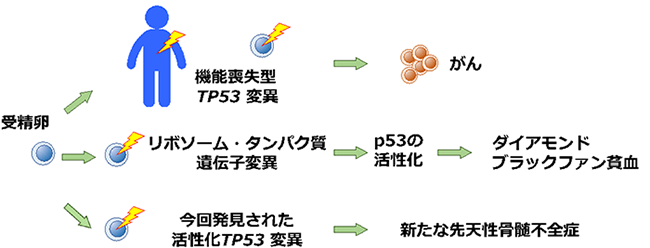
先天性骨髄不全症は、血液を作り出す細胞が先天的に障害され、赤血球や白血球などが減少してしまう病気の総称です。その中に、赤血球の産生だけが障害され、生まれた時から重い貧血に悩まされるダイアモンド・ブラックファン貧血(DBA)※1という病気があります。伊藤教授らは10年ほどかけて日本における本症の原因遺伝子の検索を進めてきましたが、今回、DBAと臨床的に診断された患者さんの中に、がん抑制遺伝子※2TP53※3の活性化変異が原因で起こる「新たな先天性骨髄不全症」を発見しました。
本研究は、”TP53の遺伝子産物p53の活性化が骨髄不全症を引き起こす” という従来の仮説を直接的に証明しました。さらに、重要ながん抑制遺伝子TP53の機能について新たな知見を見出し、先天性骨髄不全症の新規治療法やがんの予防法の開発に結びつく可能性を提供するものです。
本研究成果は、米国学術雑誌「The American Journal of Human Genetics」に掲載されました。(オンライン版公開日:日本時間 2018年8月24日 午前1時)
TP53遺伝子と疾患
がんを高頻度に発症するリ・フラウメニ症候群では、がん抑制遺伝子TP53の機能喪失型変異が認められます。一方、ダイアモンド・ブラックファン貧血(DBA)ではその遺伝子産物p53の活性化が重要な役割を果たしていると考えられています。今回、DBAと臨床的に診断された患者さんの中に、TP53の活性化変異が原因で起こる「新たな先天性骨髄不全症」を発見しました。
なお、本研究は、京都大学iPS細胞研究所の江藤浩之教授、筑波大学医学医療系生命医科学域の高橋智教授、宮崎大学フロンティア科学実験総合センターの剣持直哉教授、東京大学医科学研究所ヒトゲノム解析センターの宮野悟教授、名古屋大学小児科の小島勢二名誉教授、山梨大学小児科の杉田完爾教授、国立成育医療センター血液内科の石黒精診療部長をはじめ、多くの方々との共同研究で、国立研究開発法人日本医療研究開発機構(AMED)難治性疾患実用化研究事業等の支援を受けて行われました。
研究の背景
新生児期から重い貧血がみられるダイアモンド・ブラックファン貧血(Diamond-Blackfan貧血、以下 DBA)※1は、貧血の他に多指症などの奇形が約40%で認められることや、将来がんや白血病を発症しやすくなるといった特徴があります。この病気には、がん抑制遺伝子※2TP53※3が重要な役割を果たしている可能性が以前から指摘されていました。TP53遺伝子の異常(変異)は、ヒトのがんで最も高い頻度で見られ、変異によりその遺伝子産物p53たんぱく質(以下、p53)が機能を失うためにがんが発生することが知られています。しかし、DBAではp53がどのような役割を果たしているか、詳細なところは明らかになっておらず、仮説として論じられて来ました。これまで報告されてきたDBAの原因遺伝子はTP53ではなく、ほとんどがリボソーム※4という細胞内複合体に関わるものです。リボソームは細胞の中でたんぱく質をつくる働きがあります。このため現在では、DBAはリボソームの機能障害で起こる疾患(リボソーム病)であると考えられています。
伊藤教授らは、10年ほどかけて180例のDBA症例の原因遺伝子を検索し、新しい原因遺伝子 RPL27、 RPS27、 RPS15A を発見してきました。しかし、日本においてはまだ40%ほどの症例で原因遺伝子が分かっていません。
研究成果の概要
伊藤教授らが調べた中に典型的なDBAとは少し異なる二人の患者さんが含まれていました。二人の患者さんは、貧血に加え、低ガンマグロブリン血症、小頭症、高度の成長障害などの共通の臨床的特徴を持っていました。本研究において、両症例にはがん抑制遺伝子TP53に変異があることを発見し、DBAに類似した新しい先天性骨髄不全症であることを明らかにしました。
遺伝的にTP53の変異が認められる病気で最も知られているのは、家族内にがんを高頻度に発症するリ・フラウメニ症候群※5で、TP53遺伝子に880種類以上の変異がみつかっています。がん細胞の中にみられる変異はさらに多く、2万9千種類以上が報告され、これらの変異ではTP53の機能が失われます。一方、2症例でみつかった変異は、これまで報告されたいずれとも異なっており、本来のTP53遺伝子が作るたんぱく質よりわずかに短くなったものでした。失われた部分は p53で機能がよく分かっていない部分でしたが、変異のあるp53の機能はむしろ活性化しており、リ・フラウメニ症候群やがんで見つかった変異と性質の異なるものでした。
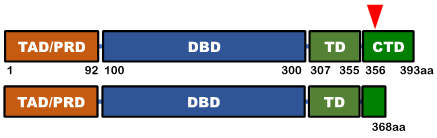
正常 p53因子(上)と患者さんに認められたp53因子(下)の比較
変異により、p53因子が短くなっています。
赤矢頭:変異が認められた部分、 TAD/PRD:転写活性化ドメイン(他のたんぱく質と結合し、転写因子としての機能を調節する領域)、DBD:DNA結合ドメイン(遺伝子のプロモーターなどDNAの特定の配列に結合する領域)、TD:四量体形成ドメイン(p53の四量体形成に関わる領域)、CTD:C末端ドメイン(機能はよく分かっていません)
本研究で用いられた手法
1)ゲノム解析:全エクソンシーケンス解析で遺伝子全体を網羅的に解析しました。
2)変異p53の転写活性への影響:今回見つかった変異が、機能喪失型かどうかをみるために、p53のよく知られた標的遺伝子CDKN1A(p21)※6への影響を解析しました。その結果、変異p53はむしろp21を発現させる力が正常p53より強いことが分かりました。
3)ゼブラフィッシュ・モデル:患者さんと類似した変異をもつゼブラフィッシュを作成しました。その結果、初期発生と赤血球造血が障害されることが明らかになりました。
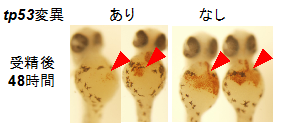
ゼブラフィッシュ・疾患モデル
変異をもつゼブラフィッシュで赤血球造血の障害が認められました。
赤矢頭:ヘモグロビン染色陽性の赤血球系細胞
4)ヒトiPS細胞モデル:ヒト細胞のゲノム編集を行い、患者さんにみられたのとほとんど同じ変異を導入しました。この細胞を赤血球へ分化誘導したところ、赤血球造血能(赤血球を作る能力)が有意に低下していることが明らかとなりました。
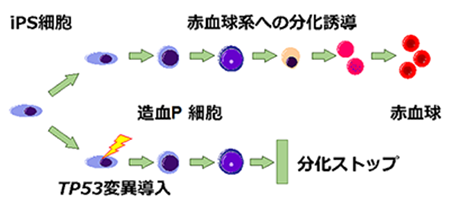
ヒトiPS細胞疾患モデル
ゲノム編集で患者さんとほとんど同じ変異を導入したヒトiPS細胞は、赤血球を作る能力が低下していました。
本研究のブレークスルーのポイントと今後の展開
極めて稀な遺伝性疾患で原因が分からないものを希少疾患と呼びます。その数は7000ほどもあり、ひとつひとつの頻度は低いのですが、全体を合わせると頻度が低いという訳ではありません。DBAは日本全体でも年間に10例ほどしか発症しませんが、今回10年に渡って全国の患者さんの変異検索をすることにより、その類縁疾患の一つをみつけることができました。本研究はp53の活性化が骨髄不全症を引き起こすことを直接的に証明しました。今回の研究成果は、先天性骨髄不全症の新規治療法やがんの予防法の開発にも結びつく可能性があります。
用語解説
- ※1 ダイアモンド・ブラックファン貧血(DBA)
- 国の指定する難病(指定難病)の一つで、全国の患者数は約250人と推定される。赤血球造血のみが障害される先天性骨髄不全症のひとつ。新生児期に貧血を発症することが多く、1歳までに90%が発症する。約半数に種々の奇形や低身長を合併する。リボソームの機能障害が、貧血を引き起こす中心的なメカニズムであると考えられおり、半数以上でリボソームたんぱく遺伝子に異常がみられる。治療は、輸血とステロイド療法が基本である。治療抵抗例では、同種骨髄移植の適応がある。生命予後は良好であるが、ステロイド療法と輸血依存症例が約40%ずつ存在しており、その副作用や合併症のために、生活の質は高いとは言えない。悪性腫瘍を合併しやすい。
- ※2 がん抑制遺伝子
- がんの発症を抑制するたんぱく質の遺伝情報をコードしている遺伝子。この遺伝子が壊れると発がんにつながることからそう呼ばれるようになった。細胞周期を制御するものや、DNA修復に関わるもの、転写を制御するものなど機能は様々である。
- ※3 TP53遺伝子
- p53遺伝子とも言われた。転写因子 p53の遺伝情報をコードしており、この転写因子は、細胞周期やDNA修復、アポトーシスなどに関わる遺伝子を活性化することにより発がんを抑制している。がんにおいて最も高頻度に変異が認められる遺伝子である。
- ※4 リボソーム
- RNA(4種類)とたんぱく質(80種類)からなる細胞内複合体。細胞内でたんぱく質の合成に関わっている。
- ※5 リ・フラウメニ症候群
- 家族性にがんを多発する遺伝症候群のひとつ。発がんリスクは30歳までに50%、60歳までに90%以上とされる。TP53の機能喪失型変異が原因である。
- ※6 CDKN1A(p21)
- サイクリン依存性キナーゼ阻害因子1遺伝子。p21(CIP1/WAF1)たんぱく質をコードしている。p21(CIP1/WAF1)は、細胞分裂の進行を調節する。p21(CIP1/WAF1)の発現は、p53によって厳密に制御されており、p53によって活性化されると、細胞分裂を止めることが知られている。
掲載論文
- 雑誌名:
- The American Journal of Human Genetics
- 論文名:
- De novo mutations activating germline TP53 in an inherited bone marrow failure syndrome(遺伝性骨髄不全症におけるTP53遺伝子の活性化生殖細胞系列変異)
- 著者名:
- 土岐力、吉田健一、王汝南、中村壮、前川貴伸、合井久美子、加藤恵美、水野聖哉、杉山文博、金崎里香、上地珠代、中島由香里、佐藤悠祐、奥野友介、佐藤亜以子、塩澤裕介、片岡圭介、白石友一、眞田昌、千葉健一、田中洋子、照井君典、佐藤知彦、神尾卓哉、坂口大俊、大賀正一、倉光球、浜口功、小原明、菅野仁、宮野悟、小島勢二、石黒精、杉田完爾、剣持直哉、高橋智、江藤浩之、小川誠司、伊藤悦朗
本研究は、国立研究開発法人日本医療研究開発機構(AMED)難治性疾患実用化研究事業①「先天性赤芽球癆(Diamond-Blackfan貧血)の新規原因遺伝子の同定と病態解明に関する研究」(研究開発代表者:伊藤悦朗)、②「稀少小児遺伝性血液疾患に対する次世代シークエンサーを利用した診断システムの開発に関する研究」(研究開発代表者:小島勢二)、③「オミクス解析技術と人工知能技術による難治性造血器疾患の病因解明と診断向上に貢献する解析基盤の開発」(研究開発代表者:宮野悟)、厚生労働科学研究費補助金難治性疾患等政策研究事業「先天性骨髄不全症の診断基準・重症度分類・診療ガイドラインの確立に関する研究」(研究代表者:伊藤悦朗)、日本学術振興会基盤研究(C)「ダイアモンド・ブラックファン貧血の新規原因遺伝子の同定と発症機構の解明」(研究代表者:土岐力)の支援を受けて行ったものです。
本研究のまとめ
- 1DBAと臨床的に診断された患者さんの中に、がん抑制遺伝子TP53の変異が原因で起こる「新たな先天性骨髄不全症」を発見しました。
- 「新たな先天性骨髄不全症」は、貧血に加え、低ガンマグロブリン血症、小頭症、高度の成長障害などの臨床的特徴があります。
- 今回見つかったTP53遺伝子の変異は、これまでがんなどで報告されている変異とは異なっており、本来のTP53遺伝子が作るたんぱく質よりわずかに短くなったものでした。この変異p53の機能はむしろ活性化しており、リ・フラウメニ症候群やがんで見つかった変異と性質の異なるものでした。
- 本研究はp53の活性化が先天性骨髄不全症を引き起こすことを直接的に証明しました。今回の研究成果は、先天性骨髄不全症の新規治療法やがんの予防法の開発にも結びつく可能性があります。
本件に関するお問い合わせ
研究に関すること
弘前大学大学院医学研究科小児科学
教授 伊藤 悦朗(いとう えつろう)
広報に関すること
弘前大学医学研究科総務グループ(総務担当)
係長 高田 光浩(たかだ みつひろ)
AMEDに関すること
国立研究開発法人日本医療研究開発機構
戦略推進部 難病研究課

