")
- 症状が現れるずっと前から、脳には多くの変化が起こっている。このことは、脳が徐々に劣化していくプリオン病に関する2つの研究において、マウスで示されている。この結果は、病気の極めて早い段階での変化を調べることができることを示唆しており、治療法を開発する上で重要である。
Many changes take place in the brain long before symptoms appear. This has been shown in mice in two studies of prion diseases in which the brain gradually deteriorates. The results suggest that changes can be studied extremely early in the disease process, which is important if we are to develop treatments.
- プリオン感染マウスでは、特定の神経集団におけるトランスラトームの変化が、脳波の変化に先行して起こる Distinct translatome changes in specific neural populations precede electroencephalographic changes in prion-infected mice
- 致死性家族性不眠症のトランスラトームプロファイリングにより、ソマトスタチンニューロンにおけるTORシグナルが関与していることが判明 Translatome profiling in fatal familial insomnia implicates TOR signaling in somatostatin neurons
症状が現れるずっと前から、脳には多くの変化が起こっている。このことは、脳が徐々に劣化していくプリオン病に関する2つの研究において、マウスで示されている。この結果は、病気の極めて早い段階での変化を調べることができることを示唆しており、治療法を開発する上で重要である。 Many changes take place in the brain long before symptoms appear. This has been shown in mice in two studies of prion diseases in which the brain gradually deteriorates. The results suggest that changes can be studied extremely early in the disease process, which is important if we are to develop treatments.
2022-10-10 スウェーデン・リンショーピング大学
プリオンとは、ある特定のタンパク質のみからなる感染性物質である。プリオンタンパク質の正常型はすべての哺乳類に存在しますが、奇妙な変化を遂げて毒性を持つようになる。この変化は、正常なタンパク質分子を毒性のある変異型に変換する変化のカスケードを引き起こす。
今回の研究では、さまざまなプリオン病の疾患モデルをマウスで観察した。研究者たちは、症状が現れるずっと前に病気のメカニズムを発見することができる新しい方法を用いた。
研究チームは、プリオンに感染したときにさまざまな種類の細胞がどのように反応するかを調べ、もう1つの研究では、致死性家族性不眠症とクロイツフェルト・ヤコブ病(CJD)という2つの遺伝性プリオン病について研究してきた。FFIは重度の不眠症が特徴で、CJDは認知症や認知機能の低下が主な症状である。この2つの病気は、後期には異なる症状を示すが、症状が現れる前の初期段階には共通点があることがわかった。
研究では、疾患のごく初期に、遺伝子発現が協調的に変化していることが確認された。
最初の研究では、病気の初期段階、つまり症状が現れる直前に非常に多くの遺伝子の発現パターンが変化していたため、治療標的を選択することは不可能であった。
2番目の研究では、プリオン遺伝子の異なる変異によって引き起こされる2つの遺伝的プリオン病について、6種類の細胞を観察した。
<関連情報>
- https://liu.se/en/news-item/skador-i-hjarnan-kan-studeras-langt-innan-symtom-uppstar
- https://journals.plos.org/plospathogens/article
- https://www.life-science-alliance.org/content/5/11/e202201530
プリオン感染マウスでは、特定の神経集団におけるトランスラトームの変化が、脳波の変化に先行して起こる Distinct translatome changes in specific neural populations precede electroencephalographic changes in prion-infected mice
Lech Kaczmarczyk,Melvin Schleif,Lars Dittrich,Rhiannan H. Williams,Maruša Koderman,Vikas Bansal,Ashish Rajput,Theresa Schulte,Maria Jonson,Clemens Krost,Fabio J. Testaquadra,Stefan Bonn,Walker S. Jackson
PLoS Pathology Published: August 12, 2022
DOI:https://doi.org/10.1371/journal.ppat.1010747
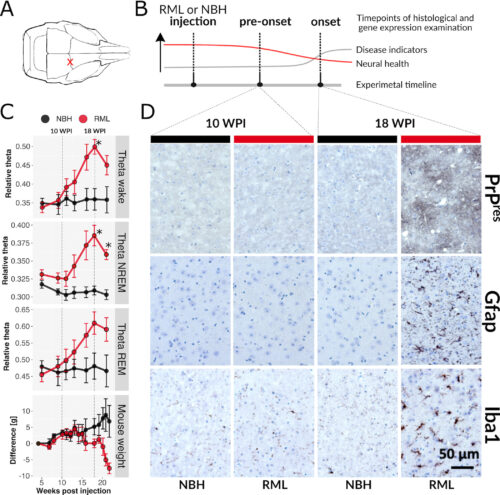
Abstract
Selective vulnerability is an enigmatic feature of neurodegenerative diseases (NDs), whereby a widely expressed protein causes lesions in specific cell types and brain regions. Using the RiboTag method in mice, translational responses of five neural subtypes to acquired prion disease (PrD) were measured. Pre-onset and disease onset timepoints were chosen based on longitudinal electroencephalography (EEG) that revealed a gradual increase in theta power between 10- and 18-weeks after prion injection, resembling a clinical feature of human PrD. At disease onset, marked by significantly increased theta power and histopathological lesions, mice had pronounced translatome changes in all five cell types despite appearing normal. Remarkably, at a pre-onset stage, prior to EEG and neuropathological changes, we found that 1) translatomes of astrocytes indicated reduced synthesis of ribosomal and mitochondrial components, 2) glutamatergic neurons showed increased expression of cytoskeletal genes, and 3) GABAergic neurons revealed reduced expression of circadian rhythm genes. These data demonstrate that early translatome responses to neurodegeneration emerge prior to conventional markers of disease and are cell type-specific. Therapeutic strategies may need to target multiple pathways in specific populations of cells, early in disease.
Author summary
Prions are infectious agents composed of a misfolded protein. When isolated from a mammalian brain and transferred to the same host species, prions will cause the same neurodegenerative disease affecting the same brain regions and cell types. This concept of selective vulnerability is also a feature of more common types of neurodegenerative diseases, such as Alzheimer’s, Parkinson’s, and Huntington’s. To better understand the mechanisms behind selective vulnerability, we studied disease responses of five cell types with different vulnerabilities in prion-infected mice at two different disease stages. Responses were measured as changes to mRNAs undergoing translation, referred to as the translatome. Before prion-infected mice demonstrated typical disease signs, electroencephalography (a method used clinically to characterize neurodegeneration in humans) revealed brain changes resembling those in human prion diseases, and surprisingly, the translatomes of all cells were drastically changed. Furthermore, before electroencephalography changes emerged, three cell types made unique responses while the most vulnerable cell type did not. These results suggests that mechanisms causing selective vulnerability will be difficult to dissect and that therapies will likely need to be provided before clinical signs emerge and individually engage multiple cell types and their distinct molecular pathways.
致死性家族性不眠症のトランスラトームプロファイリングにより、ソマトスタチンニューロンにおけるTORシグナルが関与していることが判明 Translatome profiling in fatal familial insomnia implicates TOR signaling in somatostatin neurons
Susanne Bauer, Lars Dittrich, Lech Kaczmarczyk, Melvin Schleif, Rui Benfeitas, Walker S Jackson
Life Science Alliance Published 3 October 2022
DOI: 10.26508/lsa.202201530

Abstract
Selective neuronal vulnerability is common in neurodegenerative diseases but poorly understood. In genetic prion diseases, including fatal familial insomnia (FFI) and Creutzfeldt–Jakob disease (CJD), different mutations in the Prnp gene manifest as clinically and neuropathologically distinct diseases. Here we report with electroencephalography studies that theta waves are mildly increased in 21 mo old knock-in mice modeling FFI and CJD and that sleep is mildy affected in FFI mice. To define affected cell types, we analyzed cell type–specific translatomes from six neuron types of 9 mo old FFI and CJD mice. Somatostatin (SST) neurons responded the strongest in both diseases, with unexpectedly high overlap in genes and pathways. Functional analyses revealed up-regulation of neurodegenerative disease pathways and ribosome and mitochondria biogenesis, and down-regulation of synaptic function and small GTPase-mediated signaling in FFI, implicating down-regulation of mTOR signaling as the root of these changes. In contrast, responses in glutamatergic cerebellar neurons were disease-specific. The high similarity in SST neurons of FFI and CJD mice suggests that a common therapy may be beneficial for multiple genetic prion diseases.
")
")