
2018-01-15 東京大学 日本医療研究開発機構
発表者
嶋田 一夫(東京大学大学院薬学系研究科 薬科学専攻 教授)
発表のポイント
- 副作用の引き金であるGPCRのC末端領域のリン酸化に伴い、C末端領域と膜貫通領域が、分子内相互作用を形成することを明らかにしました。
- この分子内相互作用により、GPCRの膜貫通領域が構造変化して、副作用のシグナルが促進されることを明らかにしました。
- 副作用を発現する状態でのみ形成されるこの分子内相互作用を標的とすることで、既存の医薬品の治療効果を維持しながら、副作用のみを軽減させる新規の医薬品開発が可能となることが期待されます。
発表概要
医薬品の30%以上は、Gタンパク質共役型受容体(以下GPCR、注1)と呼ばれる膜タンパク質に作用し、治療効果を発揮します。しかし、同時に副作用が生じることもあります。これは、医薬品がGPCRを介して治療効果のシグナルと副作用のシグナルの両方を細胞内に流してしまうことに起因します。したがって、GPCRが副作用のシグナルを流してしまう状態を抑えることができれば、副作用の軽減に有効です。しかし、これまでの研究では、治療効果および副作用を発揮するGPCRでどのような構造上の違いがあるかは不明でした。
東京大学大学院薬学系研究科および次世代天然物化学技術研究組合の嶋田一夫教授のグループは、溶液核磁気共鳴法(NMR法、注2)を用いて、副作用のシグナルを流す状態に対応するGPCRが、膜貫通領域やリン酸化されたC末端領域が特徴的な構造を取ることを解明しました。本研究により、副作用の発現に必要な構造の形成を阻害する薬剤の設計が可能になり、副作用を軽減した医薬品の開発が加速することが期待されます。
東京大学大学院薬学系研究科および次世代天然物化学技術研究組合の嶋田一夫教授のグループは、溶液核磁気共鳴法(NMR法、注2)を用いて、副作用のシグナルを流す状態に対応するGPCRが、膜貫通領域やリン酸化されたC末端領域が特徴的な構造を取ることを解明しました。本研究により、副作用の発現に必要な構造の形成を阻害する薬剤の設計が可能になり、副作用を軽減した医薬品の開発が加速することが期待されます。
発表内容
GPCRは、市販される医薬品の標的分子の30%を占める、創薬標的として極めて重要な膜タンパク質ファミリーです。GPCRを標的とする医薬品は、GPCRを介して、細胞内のGタンパク質を活性化する経路と、GPCRキナーゼによる自身のリン酸化を経てアレスチンを活性化する経路の、2つのシグナル経路を制御して薬理作用を発揮します(図1)。2つのシグナル経路の一方が医薬品の治療効果を、もう一方が副作用を誘起することが報告されており、例えば鎮痛薬の標的GPCRであるオピオイド受容体では、Gタンパク質の経路が鎮痛作用を誘起する一方で、アレスチンの経路が副作用に相当する耐性・依存性の発現に関与します。このことから、副作用のシグナル伝達のみを阻害することで副作用を軽減する医薬品の開発が試みられています。このような医薬品を合理的に設計するためには、GPCRが副作用を起こす際に特徴的な構造を調べることが必要です。しかし、これまで治療効果を発揮する状態と副作用を発現する状態におけるGPCRの構造の違いは見出されていませんでした。
東京大学大学院薬学系研究科および次世代天然物化学技術研究組合の嶋田一夫教授のグループは、代表的なGPCRであるβ2アドレナリン受容体(以下β2AR)を解析対象として、核磁気共鳴(NMR)を用いて、副作用発現の第一段階に対応するC末端領域のリン酸化を受けた状態、およびアレスチンが結合した状態における構造を解析しました。その結果、従来は特定の構造をとらないと考えられていたC末端領域が、リン酸化に伴ってβ2ARの膜貫通領域の細胞内側と相互作用することが明らかとなりました(図2)。加えて、上記の相互作用を経てβ2ARの膜貫通領域の構造が変化することが分かりました。さらに、この構造が、アレスチンが結合した状態の構造と類似していることが判明しました(図2)。
以上の結果は、リン酸化されたGPCRの細胞内領域で形成される分子内相互作用が、アレスチンの結合およびそれに続く副作用を発現するシグナルを活性化することを示しています(図2)。この分子内相互作用を阻害するような化合物は、細胞外領域のポケットに結合する従来型の医薬品と併用することで、治療効果の発現を維持しながら副作用のみを軽減させる効果を持つと考えられます。このようなメカニズムを持つ医薬品はこれまでに例がなく、GPCRを標的とした新しい医薬品開発を加速させることが期待されます。
※この研究は、日本医療研究開発機構(AMED)次世代治療・診断実現のための創薬基盤技術開発事業「天然化合物及びITを活用した革新的医薬品創出技術」、経済産業省の研究補助金によって行われました。
東京大学大学院薬学系研究科および次世代天然物化学技術研究組合の嶋田一夫教授のグループは、代表的なGPCRであるβ2アドレナリン受容体(以下β2AR)を解析対象として、核磁気共鳴(NMR)を用いて、副作用発現の第一段階に対応するC末端領域のリン酸化を受けた状態、およびアレスチンが結合した状態における構造を解析しました。その結果、従来は特定の構造をとらないと考えられていたC末端領域が、リン酸化に伴ってβ2ARの膜貫通領域の細胞内側と相互作用することが明らかとなりました(図2)。加えて、上記の相互作用を経てβ2ARの膜貫通領域の構造が変化することが分かりました。さらに、この構造が、アレスチンが結合した状態の構造と類似していることが判明しました(図2)。
以上の結果は、リン酸化されたGPCRの細胞内領域で形成される分子内相互作用が、アレスチンの結合およびそれに続く副作用を発現するシグナルを活性化することを示しています(図2)。この分子内相互作用を阻害するような化合物は、細胞外領域のポケットに結合する従来型の医薬品と併用することで、治療効果の発現を維持しながら副作用のみを軽減させる効果を持つと考えられます。このようなメカニズムを持つ医薬品はこれまでに例がなく、GPCRを標的とした新しい医薬品開発を加速させることが期待されます。
※この研究は、日本医療研究開発機構(AMED)次世代治療・診断実現のための創薬基盤技術開発事業「天然化合物及びITを活用した革新的医薬品創出技術」、経済産業省の研究補助金によって行われました。
発表雑誌
- 雑誌名:Nature Communications(2018年1月15日オンライン版)
- 論文タイトル:Phosphorylation-induced conformation of β2-adrenoceptor related to arrestin recruitment revealed by NMR
- 著者:Yutaro Shiraishi, Mei Natsume, Yutaka Kofuku, Shunsuke Imai, Kunio Nakata, Toshimi Mizukoshi, Takumi Ueda, Hideo Iwaï, Ichio Shimada*
- DOI番号:10.1038/s41467-017-02632-8
用語解説
- (注1)Gタンパク質共役型受容体、GPCR
- 7回膜貫通型の構造を特徴とする膜タンパク質ファミリー。細胞膜上に発現し、細胞外からのリガンド刺激に応答して、細胞内のシグナル伝達分子を活性化させることにより細胞応答を誘起する。医薬品の最大の標的分子として認識されており、現在市販されている医薬品の多くがGPCRを標的としている。
- (注2)核磁気共鳴法、NMR法
- 静磁場中に置かれた原子核の共鳴を観測する分光法。溶液中のタンパク質などの分子の構造や運動性の性質を分子レベルで調べることができる。
添付資料

図1. GPCRのシグナル伝達の模式図。鎮痛薬の標的GPCRでは、細胞内のGタンパク質を活性化するシグナルは鎮痛作用などの治療効果を、GPCRキナーゼによるリン酸化を経てアレスチンを活性化するシグナルは耐性発現などの副作用を誘起する。
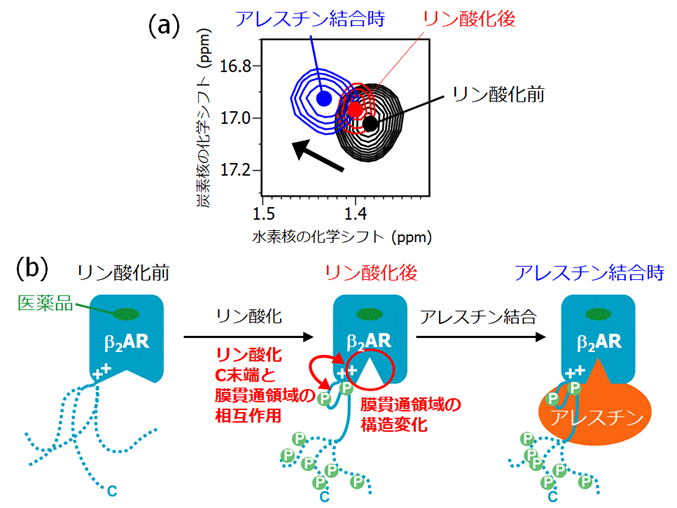
図2. (a) 膜貫通領域の構造を代表するNMRシグナル。リン酸化によってNMRシグナルがアレスチン結合時に近づいていることから、膜貫通領域の構造がアレスチン結合に有利な状態となっていると考えられる。
(b) 副作用の発現に至るまでのGPCRの構造変化。通常は特定の構造をとらないC末端領域が、リン酸化に伴い膜貫通領域と相互作用する。また、この相互作用に伴い膜貫通領域が構造変化することで、アレスチンの結合を促進する。
お問い合わせ先
内容に関するお問い合わせ
東京大学 大学院薬学系研究科 薬科学専攻
教授 嶋田 一夫(しまだ いちお)
AMEDの事業に関するお問い合わせ
日本医療研究開発機構 戦略推進部 医薬品研究課

