2019-04-12 理化学研究所
理化学研究所(理研)生命医科学研究センター骨関節疾患研究チームの池川志郎チームリーダー、郭龍研究員らの研究チームは、骨と脳を侵す新たなタイプの難病の原因遺伝子CSF1Rを発見し、CSF1R[1](コロニー刺激因子1受容体)の機能喪失が、広範かつ多様な骨格、脳神経系の異常を引き起こすことが分かりました。
本研究成果は、希少遺伝病・難病の治療法の開発につながると期待できます。
骨関節疾患研究チームは、全世界から原因遺伝子が未知の骨関節の難病の臨床データを収集しています。その中に、新たな症候群であると考えられる、共通の特徴的な骨格、脳神経系の異常を持つ3家系7人の患者を発見しました。
そこで、次世代シーケンサー[2]を用いた全エクソームシーケンス解析[3]、全ゲノムシーケンス解析[4]により、この3家系の遺伝子変異を探索・解析した結果、7人の患者全員がCSF1R遺伝子の両方の対立遺伝子座位(アレル)[5]に遺伝子の機能を喪失するタイプの塩基変化を持つことが分かりました。CSF1R遺伝子の片方のアレルの変異は、神経軸索スフェロイド形成を伴う遺伝性びまん性白質脳症(HDLS)[6]という遅発性の神経変性疾患を引き起こしますが、CSF1Rがヒトの骨格に与える異常については分かっていませんでした。CSF1Rは骨では破骨細胞[7]に、中枢神経ではミクログリア[8]に強く発現していることから、これらの細胞の機能不全が、この新しい症候群の多彩な表現型を作り出していると考えられます。
本研究は、米国人類遺伝学会の機関誌『American Journal of Human Genetics』5月号の掲載に先立ち、オンライン版(4月11日付け:日本時間4月12日)に掲載されます。

図 CSF1R遺伝子の機能喪失変異によって起こる骨と脳を犯す新たなタイプの難病の表現型
※研究支援
本研究は、日本医療研究開発機構(AMED)の難治性疾患実用化研究事業「希少難病の高精度診断と病態解明のためのオミックス拠点の構築」、日本学術振興会(JSPS)科学研究費補助金若手研究B「早期発症の骨系統疾患の原因遺伝子の系統的シーケンス解析(研究代表者:王錚)」、RIKEN-MOST Chinaによる支援を受けて行われました。
背景
骨や関節をはじめとする運動器には、多くの遺伝性難病があります。池川志郎チームリーダーらは、骨関節の遺伝性難病の原因遺伝子の同定を出発点に、その画期的な診断法・治療法の開発を目指しています。また、これら原因遺伝子の機能解析を通じて、骨格の成長・発達・維持のメカニズムを解明することも目標の一つです。そのために、全世界の医師・研究者と協力し、原因遺伝子が未知の骨関節の難病の臨床データ、画像データ(X線写真、CTなど)を収集・解析し、表現型の解明、原因遺伝子のゲノム解析などの多面的研究を行っています。これまで、29疾患の原因遺伝子を世界に先駆けて発見しました注1)。
研究チームは、最近収集した難病患者の中に、共通の骨格、脳神経系の異常を持つ3家系7人の患者を発見しました。骨格には、全身の骨硬化(骨の濃度の上昇)、脊椎の形成異常、長管骨と短管骨の骨幹端部の拡大などの特徴的な異常が、脳には、脳室(脳内部にある脳脊髄液で満たされている空間)周囲の石灰化を伴う白質脳症[9]様の神経変性とダンディ・ウォーカー奇形[10]などの脳奇形が認められました(図1)。このような骨格、脳神経系の異常の組み合わせは過去に報告がなく、新たな症候群であると考えられました。そこで、研究チームはその原因遺伝子の同定を試みました。
研究手法と成果
研究チームは、全エクソームシーケンス解析、全ゲノムシーケンス解析などの次世代シーケンサーを駆使した大規模シーケンス解析により、この3家系(家系A、B、C)の遺伝子変異を探索しました。その結果、7人の患者全員がCSF1R遺伝子の両方のアレルに変異と思われる塩基変化を持つことが分かりました。CSF1Rはコロニー刺激因子1(CSF1)の受容体で、細胞膜上に存在します。CSF1-CSF1Rを介するシグナルは、免疫に関わる単球/マクロファージ系列[11]の細胞の生存・分化・増殖に関与するとされています。単球/マクロファージ系列の細胞は、脳ではミクログリア、骨では破骨細胞です。
CSF1の変異が見つかった3家系の内の家系Aの患者は、ナンセンス変異[12]c.1441C>T(p.Gln481*)①とミスセンス変異[13]c.395C>T(p.Pro132Leu)②の複合ヘテロ接合体[14]、家系Bの患者は、イントロン変異[15]c.1859-119G>A(p.S620C_T621ins39)③と1アミノ酸の欠失変異c.1879_1881del(p.Lys627del)④の複合ヘテロ接合体、家系Cの患者は、イントロン変異c.1969+115_1969+116delAG(p.P658Sfs*24)⑤のホモ接合体[16]でした。
次に、これら①から⑤の五つの変異の機能解析を行ったところ、いずれも遺伝子の機能を喪失する変異であることが分かりました。①と⑤では、いずれもナンセンス変異により、CSF1R遺伝子のメッセンジャーRNA(mRNA)の分解が起こり、CSF1Rの発現量が著しく低下しました。また②③④では、CSF1-CSF1Rを介するシグナルの指標であるJNK[17]のリン酸化が低下し、CSF1Rの機能が低下することが分かりました(図2)。
CSF1R遺伝子の片方のアレルのみの変異は、神経軸索スフェロイド形成を伴う遺伝性びまん性白質脳症(HDLS)という常染色体優性遺伝[18]性の遅発性の神経変性疾患を引き起こします。しかし、CSF1R遺伝子の両方のアレルの変異がどのような疾患となるのかは、これまで分かっていませんでした。マウスでは、CSF1R遺伝子の機能喪失は骨濃度の上昇をもたらしますが、CSF1Rがヒトの骨格ではどのような異常を起こすのかは分かっていませんでした。
今回、血球系の細胞の受容体として知られていたCSF1Rの機能喪失が、広範かつ多様な骨格、脳神経系の異常を持つ、新たなタイプの難病の原因となることが明らかになりました。骨格の異常は、これまでに知られていた骨・関節の遺伝病の異骨硬化症[19]、もしくはPyle病(骨幹端異形成症)[20]に類似しています。脳の異常はHDLSと同様ですが、より早期に発症し、重篤で、かつHDLSでは見られない、先天的な奇形を伴っていました。
またCSF1Rは、骨では骨の吸収に重要な役割を果たす破骨細胞に、中枢神経では神経細胞の機能維持・再生に重要な役割を果たすミクログリアに強く発現します。したがって、これらの細胞の機能不全が、この新しい症候群の多彩な表現型を作り出していると考えられます。
今後の期待
次世代シーケンサーを駆使した大規模シーケンス解析は、原因が分からない希少遺伝病・難病の原因遺伝子の発見に威力を発揮しました。同様のアプローチにより、今後も多くの原因遺伝子が発見され、その遺伝子の変異の機能解析により、希少遺伝病・難病の病態が明らかになり、治療法の開発へと進むと期待できます。
また、患者の表現型の長期的な詳しい観察と患者検体の機能解析により、ヒトでのCSF1R遺伝子の機能、CSF1-CSF1Rシグナルの役割の解明が進展すると考えられます。
原論文情報
Long Guo, Débora Romeo Bertola, Asako Takanohashi, Asuka Saito, Yuko Segawa, Takanori Yokota, Satoru Ishibashi, Youichiro Nishida, Guilherme Lopes Yamamoto, José Francisco da Silva Franco, Rachel Sayuri Honjo, Chong Ae Kim, Camila Manso Mussoa, Margaret Timmons, Amy Pizzino, Ryan Taft, Bryan Lajoie, Melanie A. Knight, Kenneth H. Fischbeck, Andrew B. Singleton, Carlos R. Ferreira, Zheng Wang, Li Yan, James Y. Garbern, Pelin O. Simsek-Kiper, Hirofumi Ohashi, Pamela G. Robey, Alan Boyde, Naomichi Matsumoto, Noriko Miyake, Jürgen Spranger, Raphael Schiffmann, Adeline Vanderver, Gen Nishimura, Maria Rita dos Santos Passos-Bueno, Cas Simons, Kinya Ishikawa, Shiro Ikegawa, “Bi-allelic CSF1R mutations cause a syndrome characterized by skeletal dysplasia of dysosteosclerosis-Pyle disease spectrum and degenerative encephalopathy with brain malformation”, American Journal of Human Genetics, 10.1016/j.ajhg.2019.03.004
発表者
理化学研究所
生命医科学研究センター 骨関節疾患研究チーム
研究員 郭 龍(ゴウ・ロン)
チームリーダー 池川 志郎(いけがわ しろう)
報道担当
理化学研究所 広報室 報道担当
補足説明
-
- CSF1R
- 主に、単球/マクロファージ系列の細胞の生存・分化・増殖に関与するサイトカインであるコロニー刺激因子1(CSF1)の受容体。CSF1はcolony stimulating factor 1の、CSF1Rはcolony stimulating factor 1 receptorの略。
-
- 次世代シーケンサー
- 従来のSangerシーケンシング法を利用した蛍光キャピラリーシーケンサーである「第1世代シーケンサー」と対比させて使われている用語。多数のDNA断片を同時並行で解析し、大量の配列を読み取ることができるDNA配列解析装置。
-
- 全エクソームシーケンス解析
- ゲノム科学の手法。ゲノムの中のタンパク質に関する情報を含むエクソン部分(ゲノム全体の約3%)とその周辺のイントロン部分を、次世代シーケンサーを使って、包括的にシーケンスして解析する。
-
- 全ゲノムシーケンス解析
- ゲノム科学の手法。全ゲノムを、次世代シーケンサーを使って、包括的にシーケンスして解析する。
-
- 対立遺伝子座位(アレル)
- 哺乳類は母親と父親から同じ遺伝子セットを持つ染色体を1組ずつ受け継ぐ。この両親から受け継いだ1対の遺伝子セットを対立遺伝子座またはアレルと呼ぶ。
-
- 神経軸索スフェロイド形成を伴う遺伝性びまん性白質脳症(HDLS)
- 神経軸索スフェロイドは神経病理学的所見で、中枢神経の軸索の腫大である。遺伝性びまん性白質脳症は、常染色体優性遺伝形式をとる遺伝性の神経の変性疾患の一つ。多くは40歳~50歳代に発症する。大脳の白質部分が特に強く侵される。物忘れや意欲低下、動作が鈍くなる、歩行障害などの症状がみられる。HDLSはhereditary diffuse leukoencephalopathy with spheroidの略。
-
- 破骨細胞
- 骨の細胞の一つで、骨を破壊(吸収)する役割を担っている。骨は常に新陳代謝が行われており、骨を吸収する破骨細胞と骨を形成する骨芽細胞によって、常に壊され新しく作り直されている。
-
- ミクログリア
- 脳内に存在するグリア細胞の一種。マクロファージと同じ系列の細胞。
-
- 白質脳症
- 脳神経の変性疾患のカテゴリーの一つ。大脳の白質部分(神経線維が多い部分)が特に強く侵される1群の疾患の総称。
-
- ダンディ・ウォーカー奇形
- 脳の奇形の一つ。第4脳室と連続した後頭蓋窩正中の嚢胞と小脳虫部の完全、あるいは部分欠損を認める。
-
- 単球/マクロファージ系列
- 白血球の分化の系列の一つ。単球(単核白血球)は造血幹細胞から分化し、骨髄で成熟し、血流に入り、約2日間血中に滞在した後、組織内に入りマクロファージになる。マクロファージは、主に組織で細菌、ウイルス、死んだ細胞などの異物を取り込む働きをする。
-
- ナンセンス変異
- ナンセンス変異は、突然変異のうち、アミノ酸に対応するコドンをストップコドンに変化させる変異。その結果、そこで分断されたタンパク質を生成するか、mRNAが分解されてしまう。いずれにせよ、かなり重大な遺伝子機能の喪失をもたらす。「c.1441C>T(p.Gln481*)」は、家系Aの患者のナンセンス変異の標準表記法による表記。c.1441C>Tは塩基の変化を示し、アミノ酸をコードする領域(“c.”で示される)の1441番目の塩基がC(シトシン)からT(チミン)へ変化したことを示す。p.Gln481*は上記の塩基の変化によって起こるタンパク質の変化を示し、481番目のアミノ酸のGln(グルタミン)がストップコドン(タンパク質合成の停止信号)に変化することを示す。
-
- ミスセンス変異
- アミノ酸のコードが変化し、本来のアミノ酸ではないアミノ酸で置き換わったタンパク質が生成される変異。遺伝子機能の喪失の程度は、置き換わったアミノ酸による。「c.395C>T(p.Pro132Leu)」は、家系Bの患者のミスセンス変異の表記。c.395C>Tは塩基の変化を示し、アミノ酸をコードする領域の395番目の塩基がCからTへ変化したことを示す。p.Pro132Leuは塩基の変化によって起こるタンパク質の変化を示し、132番目のアミノ酸のPro(プロリン)がLeu(ロイシン)に変化することを示す。
-
- 複合ヘテロ接合体
- (ある遺伝子の)二つの対立遺伝子それぞれに異なる変異を持つ個体のこと。
-
- イントロン変異
- 遺伝子のmRNAに転写される部分をエキソン(exon)、エキソンとエキソンの間にあり、mRNAに転写されない部分をイントロン(intron)と呼ぶ。イントロンの変異は、mRNAのスプライシングに異常を起こすものが多い。c.1969+115_1969+116delAG(p.P658Sfs*24)は、アミノ酸をコードする領域の1969番目の塩基から数えて、115番目のA(アデニン)と116番目のG(グアニン)、2つのイントロン内の塩基が欠失(”del”)する塩基の変化が起こり、その結果、P658番目のP(プロリン)がS(セリン)に代わり、その後、フレームシフト(“fs”)が起こって、24番目にストップコドンをもつタンパク質の変化が起こったことを示す。
-
- ホモ接合体
- ある遺伝子の二つの対立遺伝子に同一の変異を持つ個体のこと。
-
- JNK
- 細胞外シグナル調節キナーゼの一つ。c-Junのアミノ酸、Ser63とSer73をリン酸化する活性を持つ。JNKはc-jun N-terminal kinaseの略
-
- 常染色体優性遺伝
- 常染色体上に存在する1対の遺伝子の一方に異常(変異)があれば発症する遺伝様式のこと。通常、患者の両親のどちらかが遺伝子変異を持つ。
-
- 異骨硬化症
- 骨系統疾患の一つ。全身の骨濃度の増加、脊椎椎体の形成不全、管状骨の骨幹端の透遼像を伴う拡大を主な特徴とする一群の常染色体劣性遺伝病の総称。これまでに、ニつの原因遺伝子、SLC39A3、TNFRSF11Aが知られている。TNFRSF11Aは、2018年に骨関節疾患研究チームが発見した(郭龍ほか J Hum Genet 2018)。
-
- Pyle病(骨幹端異形成症)
- 骨系統疾患の一つ。管状骨の骨幹端の異形成を特徴とする常染色体劣性遺伝病。原因遺伝子としてSFRP4が知られている。
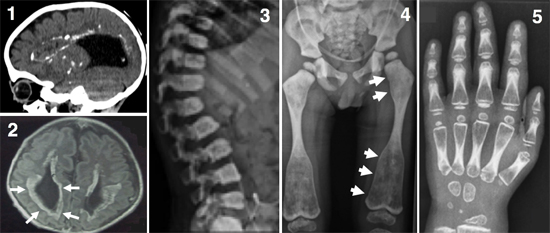
図1 骨と脳を侵す新たなタイプの難病の表現型
1:脳のCT像(側面像)。脳室の拡大(脳内黒い部分)と脳内の多数の石灰化(白い部分)が見られる。
2:脳のMRI像(冠状断)。脳の萎縮、脳室周囲の神経変性を示す高信号領域(白矢印)が見られる。
3:脊椎椎体のX線像。椎体の扁平化と硬化が見られる。
4:骨盤・下肢のX線像。骨の濃度の増加が見られ、大腿骨骨幹端部の拡大(白矢印)が特徴的である。
5:手の骨のX線像。骨の濃度の増加が見られる。
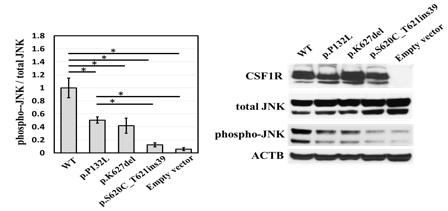
図2 変異によるCSF1R機能の低下
右は、タンパク質の発現を調べるWestern blotの実験の代表例。特異的な抗体により検出されたそれぞれのタンパク質が黒いバンドとして見える。左は、右の実験で得られたデータを基にJNKの総量(total JNK)に対するリン酸化されたJNK(phospho-JNK)の割合を計算したもの。3回行った右の実験の平均値。バーは標準偏差、*は有意水準がP<0.05であることを示す。p.P132L、p.K627del、p.S620_T621ins39は、それぞれc.395C>T ②、c.1879_1881del ④、c.1859-119G>A ③の変異によって生じる異常なタンパク質である。ACTB(ベータアクチン)は、タンパク質の発現のコントロール。CSF1-CSF1Rのシグナルは、phospho-JNKによって伝達されるため、phospho-JNKの割合が高いほど、シグナルがよく伝わっていることになる。上記三つの変異CSF1R由来のタンパク質ではいずれも、WT(野性型:変異のないCSF1Rタンパク)に比べて、JNKのリン酸化が低下していることが分かる。

の公表について")