2020-04-07 理化学研究所,日本医療研究開発機構
理化学研究所(理研)生命機能科学研究センターバイオインフォマティクス研究開発チームの笹川洋平上級研究員、田中かおりテクニカルスタッフI(研究当時)、林哲太郎技師、二階堂愛チームリーダーの研究チーム※は、「高出力型1細胞RNA[1]シーケンス法[2]」の国際的な性能比較研究に参加し、同研究室で開発された手法「Quartz-Seq2[2]」が世界最高性能を示しました。
本手法は、今後、細胞分化や臓器・器官発生などの基礎研究から、再生医療や創薬などさまざまな研究分野の発展に貢献すると期待できます。
近年、高出力型1細胞RNAシーケンス法により、臓器に含まれる全ての細胞種と機能を同定し、これにより疾患の解明や発生の機序を理解する研究が盛んに行われています。現在、ヒトの全種類の細胞を調べる国際プロジェクト「ヒト細胞アトラス(HCA)[3]」計画が進められおり、疾患解明や創薬研究などが進展すると期待されています。しかし、1細胞RNAシーケンス法はいくつかの手法が提案されており、その特性の違いが理解されていませんでした。
今回、1細胞RNA-seq法の中で世界的に主要な13手法の開発者・企業が参加し、その性能を比較する研究が実施されました。その結果、研究チームが開発したQuartz-Seq2が総合的な性能スコアで世界最高成績を収めました。
本研究は、科学雑誌『Nature Biotechnology』の掲載に先立ち、オンライン版(4月6日付:日本時間4月7日)に掲載されます。
- ※研究チーム
-
- 理化学研究所 生命機能科学研究センター
- バイオインフォマティクス研究開発チーム
- 上級研究員 笹川 洋平 (ささがわ ようへい)
- テクニカルスタッフI(研究当時) 田中 かおり(たなか かおり)
- 技師 林 哲太郎 (はやし てつたろう)
- チームリーダー 二階堂 愛 (にかいどう いとし)
本研究は、本研究チームを含むスペインのHolger Heyn博士らを中心とした25の研究機関・企業の国際共同研究グループで実施されました。
- 研究支援
- 本技術開発の研究は、科学技術振興機構(JST)戦略的創造研究推進事業(CREST)「[1細胞]統合1細胞解析のための革新的技術基盤(研究統括:菅野純夫)」の研究課題「臓器・組織内未知細胞の命運・機能の1細胞オミクス同時計測(研究代表者: 二階堂愛)」、JSTおよび日本医療研究開発機構(AMED)再生医療実現拠点ネットワークプログラム「iPS・分化細胞集団の不均質性を1細胞・全遺伝子解像度で高速に測定する技術の開発(研究代表者: 二階堂愛)」「超多検体オミクスによる細胞特性の計測(研究代表者: 二階堂愛)」の支援を受けました。また、日本学術振興会(JSPS)科学研究費補助金若手研究B「多数の1細胞から定量性のある網羅的遺伝子発現情報を取り出すための基盤技術の開発(研究代表者:笹川洋平)」の支援を受けました。
背景
私たちの臓器では、多種多様な細胞が互いに関連して臓器の機能を支えています。しかし、臓器に含まれる細胞種やその数、それぞれの細胞の機能については、十分に理解が進んでいません。臓器の疾患の理解や診断、創薬を精緻に行うためには、それらを調べる必要があります。
細胞が持つ多様な機能は、ゲノム[1]DNAから転写される数万種類のRNAの組み合わせによって決まります。RNAは、さまざまなタンパク質に翻訳され、細胞のさまざまな機能を担います。従って、臓器を構成する細胞種を判別し、その機能を類推するには、臓器に含まれている個々の細胞からRNAの量と種類を知る必要があります。これを実現する技術が、「高出力型1細胞RNA-seq法」です(図1)。二階堂チームリーダーらは、これまで高出力型1細胞RNA-seq法「Quartz-Seq2」注1, 2, 3)や1細胞完全長トータルRNAシーケンス法RamDA-seq注4, 5)などの実験技術と、その大規模なデータの解析技術注6,7)の研究で世界をリードしてきました。

図1 臓器の成り立ちや疾患の理解に貢献する高出力型1細胞RNA-seq法の概念図ヒトの体は数十兆個の細胞からなり、数百種類の細胞で構成されているが、臓器の維持に関わる幹細胞は臓器にわずかしかない。このような多様・希少な細胞の機能や状態を調べれば、臓器の成り立ちや疾患を理解できる。しかし、1細胞単位ではなく、臓器単位の実験では、希少な細胞の情報が薄まってしまい、細胞の詳しい機能までは分からない。また、多様な細胞種が混合しているため、細胞種ごとの情報も得られない。そのため、高出力型1細胞RNA-seq法を利用し、臓器を構成する一つ一つの細胞を大量に、高速かつ正確に計測する必要がある。従来法では、多くの細胞を実験できるが、検出できる遺伝子数に限界がある。そのため、細胞種類の同定は得意でも、細胞周期などの細胞状態の違いや細胞の持つさまざまな機能の同定が不得手だった。Quartz-Seq2は検出できる遺伝子数が多いため、細胞状態や機能の正確な同定が容易である。
高出力型1細胞RNA-seqの登場により、国際共同プロジェクト「ヒト細胞アトラス(HCA)」が進められています。HCAでは、ヒト細胞全種類の1細胞RNAの種類と量を収集し全細胞アトラス[4]の構築が試みられており、全細胞アトラスの完成によって、疾患の解明や創薬の研究などが進展すると期待されています。2020年3月現在、HCAには71カ国1,048の研究機関が参加しており、日本からは、本研究チームや理研生命医科学研究センターなど注8)が参加しています。
しかし、高出力型1細胞RNA-seq法の開発競争は激しく、いくつかの手法が提案されています。国際共同プロジェクトで、各国が分担してデータを収集するには、それらの特性を定量的に理解しておく必要があります。そのためには、各国の開発者や開発企業が、全く同じサンプルを対象に、各手法で実験を実施し、全く同じデータを解析する手法で公平に比較する研究が必要でした。
注1) Yohei Sasagawa, Hiroki Danno, Hitomi Takada, Masashi Ebisawa, Tetsutaro Hayashi, Akira Kurisaki, Itoshi Nikaido. “Quartz-Seq2: a high-throughput single-cell RNA-sequencing method that effectively uses limited sequence reads”. Genome Biology. 2018., doi: 10.1186/s13059-018-1407-3
注2)2013年7月25日理化学研究所プレスリリース「細胞1個の遺伝子発現を網羅的に定量化する「Quartz-Seq法」を開発」
注3)2018年3月31日理化学研究所プレスリリース「数千個の1細胞からRNA量と種類を正確に計測」
注4)2018年2月14日理化学研究所プレスリリース「1細胞から多種多様なRNAのふるまいを計測」
注5)2019年9月17日理化学研究所プレスリリース「1細胞 RNA 解析キットの商用化へ」
注6)2019年2月25日プ理化学研究所レスリリース「高速検索エンジン「CellFishing.jl」を開発」
注7)2020年1月20日理化学研究所プレスリリース「大規模データに対する主成分分析の性能を評価」
注8)2017年12月20日理化学研究所プレスリリース「アジア初のHuman Cell Atlas会議が開催されました」
研究手法と成果
高出力型1細胞RNA-seq法を公平に比較するため、その性能の国際的な比較(ベンチマーキング)研究が、スペインのHolger Heyn博士を中心としたドイツ、スウェーデン、英国、米国、本研究チームなどの25の研究機関・企業の国際共同研究チームで実施されました。また、このベンチマーキングには、主要な13手法の開発者や開発企業、実験施設などのチームが参加しました。
まず、Heyn博士らは、複数の細胞種を一定の比率で混合した細胞の混合液を作製しました。これを複数の容器に等分し、世界中の開発者、開発企業に凍結して輸送しました(図2)。各参加者は、それぞれの手法で1細胞RNA-seq法を実施し、その未加工のデータをスペインのHeyn博士らに送り返します。そして、Heyn博士らは、集められたデータを用いて、検出した遺伝子[1]数、細胞特異的マーカー遺伝子検出、細胞の分類のしやすさ、細胞型の同定、統合後の細胞分類のしやすさ、他手法との統合のしやすさの6項目の性能を定量化しました。これにより、全く同じ検体で、全く同じデータ解析手法で、公平にその性能を比較できるデータがそろいました。細胞の凍結輸送などは実際の研究で行われるプロセスを模しており、実務的なベンチマーク研究となりました。
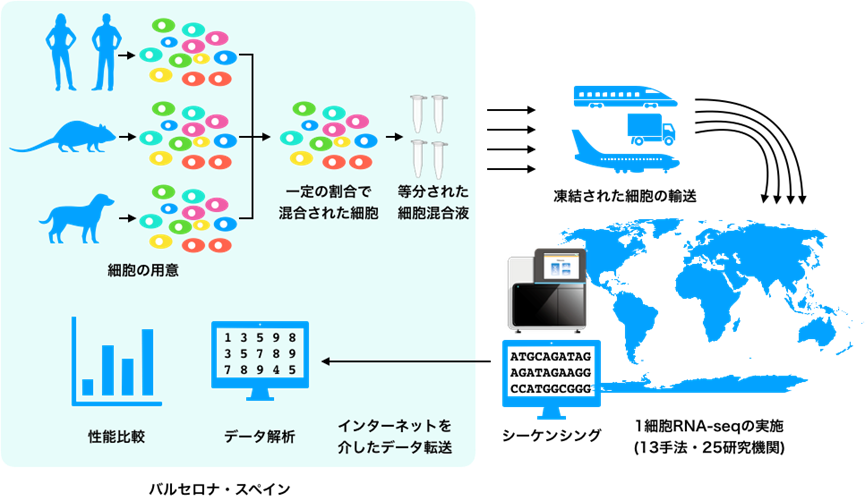 図2 国際的なベンチマーク研究の概要スペインのバルセロナにある研究所でHeyn博士らによって混合された細胞を研究に参加した各国の開発者らに配布。各拠点で1細胞RNA-seqを実施して未加工のデータをHeyn博士らのチームに転送。Heyn博士らのチームでデータ解析や性能比較を実施した。
図2 国際的なベンチマーク研究の概要スペインのバルセロナにある研究所でHeyn博士らによって混合された細胞を研究に参加した各国の開発者らに配布。各拠点で1細胞RNA-seqを実施して未加工のデータをHeyn博士らのチームに転送。Heyn博士らのチームでデータ解析や性能比較を実施した。
本研究チームは、2018年に発表したQuartz-Seq2を用いて、この性能評価研究に参加しました。その結果、研究チームが開発したQuartz-Seq2は多くの評価指標において高い成績を示しました。さらに総合的な性能スコアで最も性能の高い手法であることが示されました(図3)。特に検出した遺伝子数は他の手法に比べて1.5~5倍程度に達し、圧倒的な性能差が見られました(図4)。その理由として、微量な標的RNA分子を漏れなくDNAシーケンス可能なcDNA[5]に変換できる点にあることが考えられます。これは、これまで研究チームが証明してきた結果と同じでした注1)。

図3 各性能指標と総合性能スコアのランキング今回の性能評価研究に参加した13手法の性能を示した図。左に各性能指標を示した。右にそれらの性能指標を統合した総合性能スコアを示した。円の大きさと色で性能を示している。円が大きく黄色に近いほど性能が良い。13手法を総合性能スコアの順に並べてあり、Quartz-Seq2は1位となった。
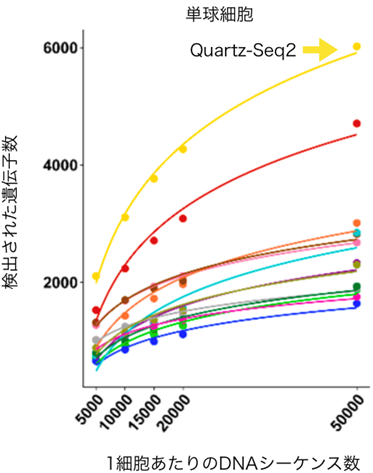
図4 検出できる遺伝子数を比較した結果各手法によってヒト単核球細胞から検出できる遺伝子の発現量を示した。横軸は1細胞あたりに割り当てられたDNAシーケンス数、縦軸は1細胞あたりの検出された遺伝子数の平均を示す。黄色の線が、Quartz-Seq2を示す。他の手法に比べて検出できる遺伝子数が1.5~5倍程度であった。一般的にシーケンス数を増やすと検出できる遺伝子数は上昇する。他の多くの手法では、いくらシーケンス数を増やしてもQuartz-Seq2で検出された遺伝子数にはたどりつかないことが分かる。
今後の期待
Quartz-Seq2には他の手法の性能を高感度化できるアイディアが含まれており、他の手法の開発にも大きく貢献すると期待できます。例えば、RNAからcDNAへの変換効率が高感度化に重要である点は、他の手法でもこの点の改善が重要であると予想されます。さらに、Quartz-Seq2で用いられている細胞ごとの反応液を混合する手法やその条件、DNA精製方法なども、ほかの方法に応用できます。
今回のベンチマーク研究と並行して、マイクロウェル型や組み合わせインデックス型など、ハイスループットな1細胞採取法[6]を用いた1細胞RNA-seq法のベンチマーキングが行われました注8)が、これらの結果でも、Quartz-Seq2の性能を上回るものはありませんでした。しかし、これらの1細胞採取法は多くの細胞を一度に採取できるため、Quartz-Seq2に比べて低感度であるものの、よりたくさんの細胞を計測することに向いています。今後、このようなハイスループットな1細胞採取法とQuartz-Seq2を組み合せると、高感度なまま、より多くの細胞を計測できる1細胞RNA-seq法が開発できる可能性があります。
高性能なQuartz-Seq2は、他の手法では捉えきれない細胞の状態の違いや細胞機能の違いを計測できます。そのため、再生医療で利用される細胞の高品質化や創薬研究、疾患の研究などに応用すれば、それらの研究がより加速されると期待できます。
論文情報
- タイトル
- Benchmarking Single-Cell RNA Sequencing Protocols for Cell Atlas Projects
- 著者名
- Elisabetta Mereu, Atefeh Lafzi, Catia Moutinho, Christoph Ziegenhain, Davis J. McCarthy, Adrian Alvarez, Eduard Batlle, Sagar, Dominic Grün, Julia K. Lau, Stéphane C. Boutet, Chad Sanada, Aik Ooi, Robert C. Jones, Kelly Kaihara, Chris Brampton, Yasha Talaga, Yohei Sasagawa, Kaori Tanaka, Tetsutaro Hayashi, Caroline Braeuning, Cornelius Fischer, Sascha Sauer, Timo Trefzer, Christian Conrad, Xian Adiconis, Lan T. Nguyen, Aviv Regev, Joshua Z. Levin, Swati Parekh, Aleksandar Janjic, Lucas E. Wange, Johannes W. Bagnoli, Wolfgang Enard, Marta Gut, Rickard Sandberg, Itoshi Nikaido, Ivo Gut, Oliver Stegle, Holger Heyn
- 雑誌
- Nature Biotechnology
- DOI
- 10.1038/s41587-020-0469-4
補足説補足説明
- [1] RNA、ゲノム、遺伝子
- 「ゲノム」は細胞が持つ全遺伝情報であり、DNA(デオキシリボ核酸)という生体高分子に4種類の塩基の配列として記録されている。ゲノムの中の遺伝情報が記録(コード)された領域が「遺伝子」である。遺伝子領域の情報は、DNAを鋳型として「RNA(リボ核酸)」が合成(転写)されることで読み出される。
- [2] 高出力型1細胞RNAシーケンス法、Quartz-Seq2
- 1細胞中に含まれるRNAをDNAシーケンサーで配列決定し、網羅的かつ定量的にその量や種類を決定する方法。微量なRNAを用いるため、微量RNAからcDNAを合成する「逆転写反応」と、シーケンス可能な量までcDNAを増幅させる「全cDNA増幅法」の二つのステップからなる。大量の1細胞由来のRNAをシーケンスできる技術を高出力型1細胞RNAシーケンスと呼ぶ。Quartz-Seq2(クォーツ・セックツー) は、数千から数万個の1細胞由来のRNAをシーケンスすることで、細胞の機能や特徴を明らかにできる計測手法。2013年に本研究チームの笹川と二階堂は高感度の1細胞RNA-seqとしてQuartz-Seqを開発した。2018年に、Quartz-Seqを基盤に、より多くの細胞由来の1細胞RNAをより高感度にシーケンスできる方法に発展させた。
- [3] ヒト細胞アトラス(HCA)
- 2016年にスタートした、ヒトが持つ全細胞種類の1遺伝子発現地図(アトラス)を計測しデータベース化する国際研究プロジェクト。HCAはHuman Cell Atlasの略。
- [4] 全細胞アトラス
- 1細胞RNA-seqを用いて、ある生物が持つ全細胞の種類の遺伝子発現をデータベース化したもの、あるいは、そのプロジェクト。数百万~数億細胞×数万遺伝子のRNA量の行列データになると見込まれる。ある生物の全細胞のデータベースを、その生物の全細胞の「地図(アトラス)」になぞらえた呼び方。
- [5] cDNA
- RNAを鋳型として逆転写酵素で合成されたDNA。RNAに対して相補鎖になるため、相補的DNA(complementary DNA)と呼ばれる。
- [6] ハイスループットな1細胞採取法
- 1細胞RNA-seqでは1細胞をそれぞれ容器に取り分けてから、細胞内のRNAを取り出してcDNAに逆転写する必要がある。一般的に1細胞の採取はセルソーターという装置と384穴のマイクロウェルプレートを利用する。384ウェルプレートを1枚採取するのに3-10分程度かかり、たくさんの細胞を1細胞RNA-seqを実施するには、この点がボトルネックになる。一方、マイクロウェル型1細胞RNA-seqは、微細加工技術を利用し、1細胞がちょうど収まる程度の穴が数万から数十万ある容器に1細胞を採取する。また組み合わせインデックス型では、細胞の混合と分離を繰り返しながら細胞を多段階的に標識することで、数万の細胞を区別して採取する方法である。これらの方法はセルソーターを利用した1細胞採取法よりも速く、たくさんの細胞を採取できる。
発表者・機関窓口
理化学研究所 生命機能科学研究センター
バイオインフォマティクス研究開発チーム
上級研究員 笹川 洋平(ささがわ ようへい)
テクニカルスタッフI(研究当時) 田中 かおり(たなか かおり)
技師 林 哲太郎(はやし てつたろう)
チームリーダー 二階堂 愛(にかいどう いとし)
生命機能科学研究センターに関する問い合わせ機関窓口
理化学研究所 生命機能科学研究センター センター長室 報道担当
山岸 敦(やまぎし あつし)
機関窓口
理化学研究所 広報室 報道担当
AMED事業に関するお問い合わせ先
日本医療研究開発機構(AMED)
再生・細胞医療・遺伝子治療事業部 再生医療研究開発課

