2022-01-07 東京大学
今野 直輝(生物科学専攻 修士課程2年生)
岩崎 渉(大学院新領域創成科学研究科 先端生命科学専攻 教授/生物科学専攻 教授(兼担))
谷内江 望(ブリティッシュコロンビア大学 准教授・Canada Research Chair/東京大学先端科学技術研究センター 客員准教授)
発表のポイント
- 大量のDNA配列から高速に巨大な系統樹を推定する「深層分散コンピューティング」FRACTAL手法(注1)を開発した。
- 本手法は、さまざまな系統樹推定ソフトウェアが取り扱えるDNA配列数を飛躍的に拡張させ、最低でも2億の配列から成る巨大な系統樹の推定に利用できることが示された。
- 従来不可能だった大規模の系譜推定を可能にし、微生物・ウイルスなどの進化過程や動物の発生過程の解明など、幅広い生物学分野における基幹手法になることが見込まれる。
発表概要
生物は、A、T、G、Cの4文字からなるゲノムDNA配列を変化させながら進化する。進化生物学では、地球上の生物のDNA配列からそれらの進化の系譜である進化系統樹を推定する技術が開発されてきた。近年、このコンセプトは発生生物学にも持ち込まれつつある。とりわけ、染色体上の部分的な人工DNA配列に高速に変異を蓄積するような仕掛けを施した受精卵から動物個体を得て、そのような動物から得た細胞一つひとつのDNAの変異情報から全身の発生過程を再構築しようというプロジェクトが世界的に進んでいる。一方で、既存の系譜推定手法は計算時間とメモリ消費量が大きく、対象とできる配列数は100万程度に限られていた。
東京大学大学院理学系研究科生物科学専攻の今野直輝修士課程学生、岩崎渉教授とブリティッシュコロンビア大の谷内江望准教授らの共同研究チームは「深層分散コンピューティング」手法FRACTALを発明し、これが2億以上の配列からなる系譜を正確に推定すること、進化系統樹推定、細胞系譜トレーシングを大きく拡張させることを示した。
本本技術によって微生物・ウイルスなどの大規模進化、動物の高解像度発生過程など、幅広い分野における生物学の地平が押し広げられることが期待される。
発表内容
生物は、A、T、G、Cの4文字からなるDNA配列を少しずつ変化させながら進化してきた。その過程では1つの種が異なる2種に分岐することが繰り返され、過去の種分化の系譜は二分岐を繰り返す「木」、すなわち進化系統樹として表現される。より類似したDNA配列を持つ二種はより最近に分岐したと想定できるため、私たちは現在地球上に生存する多数の生物種のDNA配列を比較することでそれらの進化系統樹を推定することができる。
この進化系統樹の推定に着想を得て、近年では動物の発生過程において細胞が分裂していく過程である細胞系譜をトレーシングする技術が開発されている。このようなアイデアでは、ゲノム編集技術(注2) を利用して、染色体に導入された短い人工DNA配列に細胞分裂とともに変異が導入されるような仕掛けが準備される。これを搭載した受精卵から動物個体を得て、そこから一つひとつの子孫細胞のもつDNAの変異情報を解析すると、進化系統樹推定と同じように、受精卵から細胞分裂がどのように進んでその動物個体を形成したのかという系譜情報を手に入れることができる。
しかし、一般にDNA配列から系統樹を推定する手法は計算時間とメモリ消費量が大きく、これまで系譜推定が可能な配列数は最大で100万程度に限られていた。地球上に生息する種数は真核生物だけで870万種と見積もられており、また昨今ゲノム、メタゲノム解析によって次々に同定の進む微生物・ウイルスなども考慮すると、今後進化生物学が取り扱う必要があるDNAの配列数はこの計算限界を大きく超えると考えられている。また動物の発生過程における細胞系譜トレーシングにおいても、脊椎動物が数百億個〜数兆個の細胞から成ることを考えると、今後計算機的な限界に直面することが予想されていた。
今回、東京大学大学院理学系研究科生物科学専攻の今野直輝 修士課程学生、岩崎渉 教授らの研究グループはブリティッシュコロンビア大学バイオメディカルエンジニアリングの谷内江望 准教授らとの共同研究チームによって、さまざまな系譜推定ソフトウェアの取り扱える配列数を拡張する「深層分散コンピューティング」手法FRACTALを開発した。共同研究チームは、通常の系譜推定ソフトウェアが逐次的に計算を進める中で取り扱う全ての配列の計算状況をモニタリングすることが、多くの実践的な課題を解くためには過剰計算であるという仮説を立て、FRACTALを実装するという形でこれを示した。
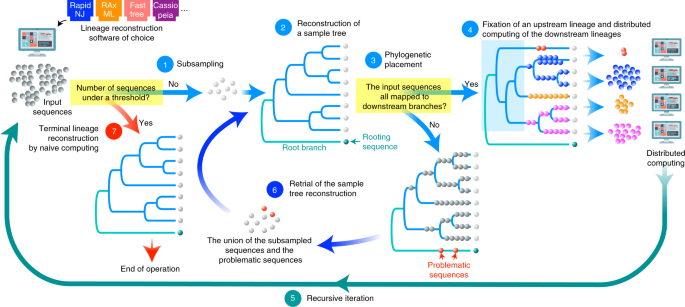
FRACTALは、任意の系譜推定ソフトウェアをプラグインとして取り込むことができる。FRACTALではまず、入力された大量のDNA配列の一部のみをランダムにサンプリングして、それらの小さな系譜の木を再構築する(小さな計算量)(図1)。

図1:FRACTALによる巨大系統樹を推定するプロセスの概要
次に、全ての入力DNA配列のそれぞれが、その小さな木の上のどの位置に一番近いのかを推定する(小さな計算量)。その結果、もしその小さな木において入力配列がマッピングされない上流部分が現れた場合には、この上流部分については「解けていた」と判断する。このとき、上流部分の木の枝にぶら下がる形で残りの入力配列がマッピングされているので、それぞれの枝にぶら下がった配列群についてはその下流の木を独立に推定すれば良いことになる。従って、この分割された配列群に対して独立した異なる計算機を用いて同じプロセスを繰り返すことができる(もしそのような「空」の上流部分が現れなかった場合は、この結果から学んだバイアスを掛けたサンプリングを上流部分が「空」になる小さな木が得られるまで繰り返す)。このように、FRACTALでは計算機が小さなタスクをこなした後に、残りのタスクを他の計算機に振り分け、タスクを振り分けられた計算機もまた別の大量の計算機にタスクを振り分けるという形を採用することで、巨大な系譜の推定を可能にした。
本研究では、本手法を多様なDNA配列データに適用し、FRACTALがさまざまな巨大系譜推定を可能にすることを示した。例えば、シミュレーションによって2億3500万本を超える変異が導入されたDNA配列を生成し、300ノードの計算機を用いてFRACTALを実行した。その結果、32時間以内に99.8%以上の精度でシミュレーションの配列生成プロセスを再現できることを示した(図2)。
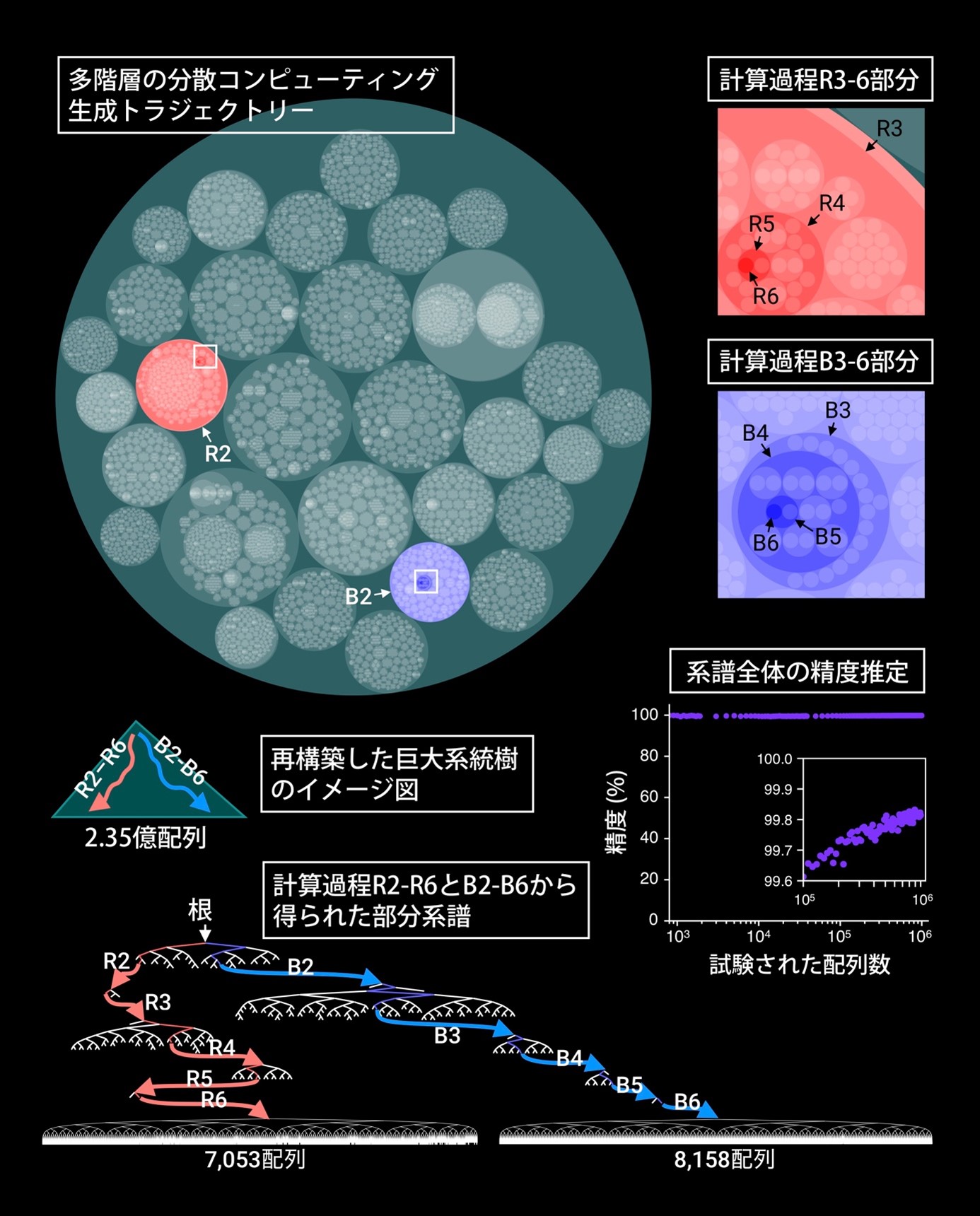
図2:FRACTALによる2.35億配列の系統樹の再構築。多階層の分散コンピューティング生成トラジェクトリーでは、一つの円が一つの計算ジョブを示し、それが生成した分散コンピューティングタスクは次の階層の円群で示される。巨大な系譜全体の精度は直接測ることができないため、部分的な系譜の精度値から精度の推定を行った。
さらに、自然界の生物のDNA配列への適用可能性を検証するため、16S rRNA遺伝子の進化から学習してシミュレーションで生成した擬似巨大進化系統樹も正確に再構築することができること、さまざまな細胞工学データを学習してシミュレーションで生成した1,600万細胞(マウスの13.5日胚の細胞数に相当)の系譜についても99.5%の精度で推定できることを示した。また、シミュレーションデータのみならず、実験的に試験管内で変異を蓄積した300万本以上のDNA配列に対してもFRACTALは妥当な系譜を推定することができた。
本手法は、今後、進化生物学、発生生物学の地平を大きく押し広げる可能性を持っている。進化生物学においては、最近は特に環境中のDNAを網羅的にシークエンシングすることで得られるメタゲノム配列の系統解析を行うことで、培養が難しい未知微生物の存在が明らかにされており、FRACTALを用いることで新しい微生物やウイルスの系統関係を網羅的に解明できる可能性がある。またFRACTALは、発生生物学分野において、さまざまな動物の全身発生過程を高解像度で解析することのできる現在唯一の計算機フレームワークとなっている。動物の全身発生地図の解明に貢献し、これが生物学におけるさまざまな新しい発見につながることが期待される。
発表雑誌
- 雑誌名
Nature Biotechnology論文タイトル
Deep distributed computing to reconstruct extremely large lineage trees著者
Naoki Konno, Yusuke Kijima, Keito Watano, Soh Ishiguro, Keiichiro Ono, Mamoru Tanaka, Hideto Mori, Nanami Masuyama, Dexter Pratt, Trey Ideker, Wataru Iwasaki, and Nozomu Yachie*DOI番号
10.1038/s41587-021-01111-2
用語解説
注1 「深層分散コンピューティング」手法FRACTAL
本研究で開発した巨大な系統樹推定のための計算手法。既存の系統樹推定ソフトウェアをプラグインとして取り込むことができ、分散コンピューティングを多階層で繰り返すことによって取り扱えるDNA配列数を大幅に拡張した。
注2 ゲノム編集
染色体上の任意の配列を正確に編集したり、ランダムな配列に変換したりする技術。2020年にノーベル化学賞がこの技術を開発したジャニファー・ダウドナ博士とエマニュエル・シャルパンティエ博士に贈られている。

