2019-06-14 理化学研究所
理化学研究所(理研)生命医科学研究センターゲノム解析応用研究チームの鎌谷洋一郎客員主管研究員、寺尾知可史チームリーダー、小杉俊一研究員らの研究チーム※は、全ゲノムシークエンス[1]データから構造多型(SV)[2]を検出する既存の69のアルゴリズム[3]の性能評価を行い、高精度でSVを検出するためのツールの選定や組み合わせの選別などを提供する基盤情報を確立しました。
本研究成果は、疾患に関わるゲノム変異の同定や個別化医療の実現に貢献するとともに、微生物、動植物を含めた全ての種のゲノムにおけるSV検出のための有用な情報を提供します。
SVは、個人間のゲノムの違いのうち50塩基対以上の長さの変異のことで、発達障害や知的障害などを含むさまざまなヒト疾患の要因になると考えられています。しかし、SVを検出する多くのアルゴリズムが開発されているにもかかわらず、精度良くSVを検出する単独のツールは存在しません。
今回、研究チームは、多くの評価データを用いて69のSV検出アルゴリズムの包括的性能評価を実施し、各SVのタイプとサイズに応じて、どのアルゴリズムが検出精度[4]や検出感度[5]などの性能が優れているかを明らかにしました。さらに、より高精度でSVを検出する手段として、アルゴリズム間で共通に検出されるSVの精度、感度を体系的に解析し、各SVのタイプとサイズに応じた、最適なアルゴリズムの組み合わせを選定するために情報基盤を確立しました。
本研究は、英国の科学雑誌『Genome Biology』(6月3日号)に掲載されました。

図 構造多型(SV)検出アルゴリズムのサイズ別欠失検出性能比較
※研究チーム
理化学研究所 生命医科学研究センター
ゲノム解析応用研究チーム
客員主管研究員 鎌谷 洋一郎(かまたに よういちろう)
チームリーダー 寺尾 知可史(てらお ちかし)
研究員 小杉 俊一(こすぎ しゅんいち)
基盤技術開発研究チーム
チームリーダー 桃沢 幸秀(ももざわ ゆきひで)
研究員 ギョウケイ・リュウ(Xiaoxi Liu)
統合生命医科学研究センター(研究当時)
副センター長 久保 充明(くぼ みちあき)
※研究支援
本研究は、日本学術振興会(JSPS)科学研究費補助金基盤研究C「低カバレッジロングリードを用いた効率的ゲノム構造変異同定手法の確立(研究代表者:小杉俊一)」による支援を受けて行われました。
背景
ゲノム「構造多型(SV)」は、50塩基対(bp)以上の欠失[6]、挿入[7]、重複[8]、逆位[9]多型の総称であり、50bpより小さい欠失、挿入に相当する「インデル」、1bpの塩基置換である「一塩基多型(SNV)」[10]とは区別されます。SVの中の欠失と重複は、コピー数多型(CNV)とも呼ばれます。その出現頻度はSNV(個人当たり~400万)やインデル(個人当たり~70万)と比較して、SV(個人当たり1万~2万)では低いものの、その大きいサイズのためにSVに起因する個人ゲノム間で観察される異なる塩基数は、SNVによる個人間の異なる塩基数の3~10倍あることが示されています。
このように個人ゲノム間に大きな違いをもたらすSVは、発達障害や知的障害などを含むさまざまなヒト疾患の遺伝的要因となることが近年の多くの研究から示されています注1,2)。また、がんなどの体細胞変異によって引き起こされる疾患においても、SVが関わることを示す多くの研究があります注3,4)。
SVの構造の複雑さと大きいサイズのために、SVの検出はSNVと比較して困難です。ゲノムの多型は通常、100bp~150bpの短い配列(リード)[11]データをヒトの標準ゲノム配列(リファレンス配列)[12]にアライメント[13]して検出します。このリード長内に収まるSNVやインデルに対して、より大きなサイズのSVはリード内に収まらず、SVをまたいでアライメントされるリードの間接的な証拠を用いて検出しなければならないため、検出精度や検出感度が低くなってしまいます。
これを克服するために、これまでに多くのSV検出アルゴリズムが開発され、コンピュータツールとして提供されてきました。しかし、それぞれのアルゴリズムを用いて得られる結果には共通性が低い問題点がありました。さらに、既存の多くのSV検出アルゴリズムを、同じ条件で一度に包括的に性能評価した研究が行われていないため、多くあるアルゴリズムからどのようなアルゴリズムを選定し、組み合わせて用いればSVを精度良く検出できるか、科学的根拠に基づいて選別できませんでした。
注1)Weischenfeldt J, et al. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet. 14, 125-38 (2013).
注2)Marshall, C.R. et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 49, 27-35 (2017).
注3)Yi, K. et al. Patterns and mechanisms of structural variations in human cancer. Exp.. Mol. Med. 50, 98 (2018).
注4)Nik-Zainal, S. et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47-54 (2016).
研究手法と成果
研究チームはまず、単独の全ゲノムシークエンスデータからSVを検出する現時点で得られるアルゴリズムのほぼ全て(79アルゴリズム)を入手しました。そして、そのうち研究チームのコンピュータ環境下で動作した69アルゴリズムの性能(検出精度、検出感度、ブレークポイント[14]同定精度、検出に要する時間とメモリー容量など)の評価を行いました。評価データとして、一つのシミュレーションデータと六つの実サンプルから得られた全ゲノムシークエンスデータを用いました。 それぞれのアルゴリズムについて、それぞれの評価データを用いてSVを検出し、検出されたSVが正解データである標準SVデータと一致(オーバーラップ)したものを正しく検出されたSVとしてカウントし、精度(Precision)と感度(Recall)を計算しました。また、精度と感度は、SVのタイプ(欠失、重複、挿入、逆位)ごとに算出し、さらに欠失と重複については、S・M・Lのサイズ別(S: < 1 Kb、M: 1~100Kb、L: > 100Kb)に算出しました。
NA12878という個人実データを用いて各アルゴリズムが検出したSVの検出性能を、SVのタイプ別にまとめた結果を図1に示します。棒の長さはF値[15]を表し、長いほど良い性能であることを意味しています。この図から、SVタイプごとに検出性能の高いアルゴリズム、低いアルゴリズムを比較して見ることができます。また、欠失と重複をサイズ別に評価すると、アルゴリズム間でサイズごとの検出性能にも差があることが分かりました(図2)。これらの結果は、他の実データを用いて得られた結果とも一致を示しました。シミュレーションデータと実データを用いた結果を統合して得られたSVタイプごと良い性能を示すアルゴリズムのリストを表1に示します。
一般的に、異なったアルゴリズムで共通に検出されたSVの精度は高いため、以前の複数の研究では、検出精度を高めるために複数のアルゴリズムから共通に検出されるSVを選別していました。しかし、アルゴリズムのどのような組み合わせで、アルゴリズム間で共通に検出されるSVの精度や感度が向上するのかを調べる体系的な解析はこれまで行われていませんでした。
そこで、SVの各タイプ、各サイズにおいて、それぞれ12~38のアルゴリズムを選定し、アルゴリズムの総当たりペア間で共通に検出されるSVの精度と感度を計測し、各アルゴリズムで用いられている六つの基本手法に基づいて集計しました。その結果、全体において、アルゴリズム間で共通に検出されるSVは、もとのそれぞれの単独アルゴリズムのものと比較して高い精度を示しましたが、検出感度は低下しました(図3)。例えば、図3aのRP(基本手法1)、RD(基本手法2)をそれぞれに持つアルゴリズム間で共通に検出されるSVは、RP(基本手法1)を基本手法として持つ単独アルゴリズムで検出されるSVの3倍の精度(青棒)を示しましたが、感度は約30%に低下しました(橙棒)。異なる基本手法をもとにしたアルゴリズム間で共通に検出されるSVは、同じ基本手法を基にしたアルゴリズム間で共通に検出されるSVよりも、全般的に高い精度を示しましたが、逆に感度は低下しました(図3)。また、アルゴリズムの組み合わせによっては、偽陰性をもたらす危険性が高くなることも同時に明らかになりました。
これらの結果から、SVの検出精度、感度を向上させるには、SVのタイプおよびサイズ別に適したアルゴリズムを選定する必要があることが示されました。さらに検出精度を向上させるためには、アルゴリズム間で共通に検出されるSVを取得する手段を得る必要がありますが、本研究成果はそのための最適なアルゴリズムの組み合わせを選定する上での有用な情報基盤を提供しています。
今後の期待
本研究成果は、ゲノムシークエンスデータからSVを高精度、高感度に検出するための情報基盤を提供するものです。これまでSVの検出は、各研究者の経験や知見をもとにアルゴリズムを選定してきましたが、今後、本研究成果をもとにそれぞれの研究目的に最適なアルゴリズムを選定でき、疾患発症の原因となるSVの検出に貢献すると考えられます。
さらに、本研究で用いられたSV検出アルゴリズム性能評価システムは、各研究者が今後新たに発表される新たなアルゴリズムの性能を正当に評価する上で、また研究者が今後新たなアルゴリズムを開発する上でも、有用な情報基盤となることが期待できます。 本研究のSV検出アルゴリズム性能評価システムは、下記URLから入手、利用することが可能です。
原論文情報
Shunich Kosugi, Yukihide Momozawa, Xiaoxi Liu, Chikashi Terao, Michiaki Kubo and Yoichiro Kamatani, “Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing”, Genome Biology, 10.1186/s13059-019-1720-5
発表者
理化学研究所
生命医科学研究センター ゲノム解析応用研究チーム
客員主管研究員 鎌谷 洋一郎(かまたに よういちろう)
チームリーダー 寺尾 知可史(てらお ちかし)
研究員 小杉 俊一(こすぎ しゅんいち)
報道担当
理化学研究所 広報室 報道担当
補足説明
-
- 全ゲノムシークエンス
- 次世代シークエンス技術または第3世代シークエンス技術を用いて、全ゲノムDNAを鋳型として配列解読をすること。この配列解読によって、全ゲノム長の数倍~数十倍の総塩基数に相当するショートリードまたはロングリードデータが生成される。構造多型の検出には、ショートリードでゲノム長の10~30倍、ロングリードで10倍以上の全ゲノムシークエンスデータを要する。
-
- 構造多型(SV)
- ゲノムの個人間の違いのうち、50bp以上の大きさの変異をいう。構造多型には、変異のパターンに応じて、欠失、挿入、重複、逆位、転座などに分類されるが、それぞれが混在した複雑なパターンを示す構造多型も存在する。通常、大きさの小さい構造多型ほど数が多いが、染色体レベルで起こる大きなサイズの構造多型も存在する。SVはStructural Variationの略。
-
- アルゴリズム
- 広く計算手法を呼ぶが、本研究では特にシークエンスデータを用いて構造多型を検出するためのコンピュータ解析ツール(ソフトウェア)、またはコンピュータ解析手法を呼ぶ。
-
- 検出精度
- 本研究では、精度は構造多型の検出正確性(Precision)を定義している。Precisionは、あるアルゴリズムが検出した構造多型のうち、正解と判断された構造多型のパーセンテージを表す。
-
- 検出感度
- 本研究では、感度は構造多型の検出効率(Recall)を定義している。Recallは、リファレンス構造多型データ(正解データ)に存在するあるタイプ(例えば、欠失)の構造多型総数のうち、あるアルゴリズムが正しく検出した構造多型のパーセンテージを表す。
-
- 欠失
- 構造多型のタイプの一つで、ゲノム配列の一部が失われた形態。挿入と並び、構造多型の中では最も多く存在する。
-
- 挿入
- 構造多型のタイプの一つで、ゲノム配列の特定の位置に別の配列が挿入された形態。挿入配列で最も多くあるものが、内在レトロ因子が挿入されたタイプで、ミトコンドリア配列やウイルスゲノム配列が挿入されたタイプもある。欠失と並び、構造多型の中では最も多く存在する。
-
- 重複
- 構造多型のタイプの一つで、ゲノム配列の一部の領域が重複して(2コピー以上)挿入された形態。欠失や挿入に比べて数は少ないが、重複された領域内に遺伝子が含まれる場合、通常の遺伝子発現パターンと異って発現されることが多いため、遺伝子機能の喪失を引き起こす欠失と同様、疾患との関わりが多く報告されている。
-
- 逆位
- 構造多型のタイプの一つで、ゲノム配列の一部が通常と逆方向に変換されている形態。構造変異のタイプの中で最も数が少ない。
-
- 一塩基多型(SNV)
- ゲノムの個人間の違いのうち、A、C、T、Gからなるヒトゲノム塩基配列上の1カ所の違い(置換)が一塩基多型と定義される。Single nucleotide variantの略。SNVのうち、ある集団内での頻度が1%以上あるものを、別にSNP(Single Nucleotide Polymorphism)と呼ぶ。
-
- リード
- DNAの配列決定(シークエンシング)によって得られるDNA断片の配列情報。次世代シークエンシング技術で得られるリードは、通常100~200bpのショートリード断片であり、ヒトゲノム解読の場合、数億~10億本のリードを得る。第3世代シークエンシング技術では、平均7~10Kb(7,000~10,000bp)のロングリードが得られる。
-
- リファレンス
- ある生物種のゲノム配列として、標準ゲノム配列として公開されているもの。ヒトでは、hg19やGRCh37などの総塩基数約3Gbのゲノムリファレンスが公開されている。リファレンスにリードデータをアライメントすることにより、標準リファレンス配列と異なるDNA多型が検出される。
-
- アライメント
- シークエンスリードをリファレンス配列上の合致する位置に対応付けすること。通常、ショートリードはbwaなどのアライメントツールを用いてアライメントし、得られたアライメントファイルを用いて構造多型を検出する。
-
- ブレークポイント
- 構造多型の始まりと終わりを示すリファレンス配列の境界位置。通常、挿入では1箇所、その他の構造多型では2箇所のブレークポイントが存在する。ブレークポイント情報によって、構造多型のゲノム上の位置と長さが分かる。
-
- F値
- 検出精度と検出感度を統合した統計値で、精度と感度が高くなるほどF値も高くなる。精度と感度の積に2を掛けた値を精度と感度の和で割った値。
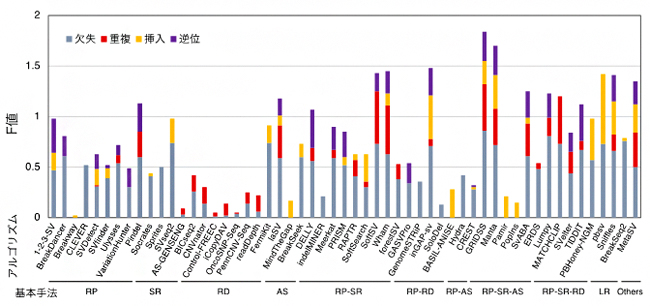
図1 構造多型(SV)検出アルゴリズムの構造多型タイプ別性能
図中に示されたアルゴリズムとNA12878実データを用いて各タイプのSVが検出され、精度(Precision)と感度(Recall)が計測された。SVタイプごとのF値(精度と感度を統合した値)が棒の長さで示される(欠失: 灰、重複: 赤、挿入: 橙、逆位: 紫)。各アルゴリズムは、基本手法(RP: read pair、SR: split read、RD: read depth、AS: assembly、LR: long read、RP-SR, RP-RD, RP-AS, PR-SR-AS, RP-SR-RD: RP, SR, RD, ASを組み合わせた手法)ごとに表示される。
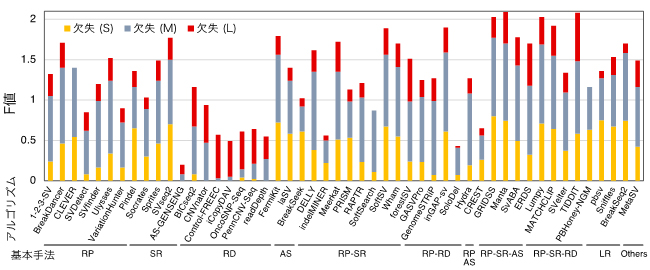
図2 SV検出アルゴリズムの欠失のサイズ別性能
図中に示されたアルゴリズムとNA12878実データを用いて各サイズ(S: < 1Kb、M: 1-100Kb、L: > 100Kb)の欠失が検出され、精度(Precision)と感度(Recall)が計測された。サイズごとのF値(精度と感度を統合した値)が棒の長さで示される(欠失 (S): 橙、欠失 (M): 灰、欠失 (L): 赤)。各アルゴリズムは、基本手法(RP: read pair、SR: split read、RD: read depth、AS: assembly、LR: long read、RP-SR, RP-RD, RP-AS, PR-SR-AS, RP-SR-RD: RP, SR, RD, ASを組み合わせた手法)ごとに表示される。
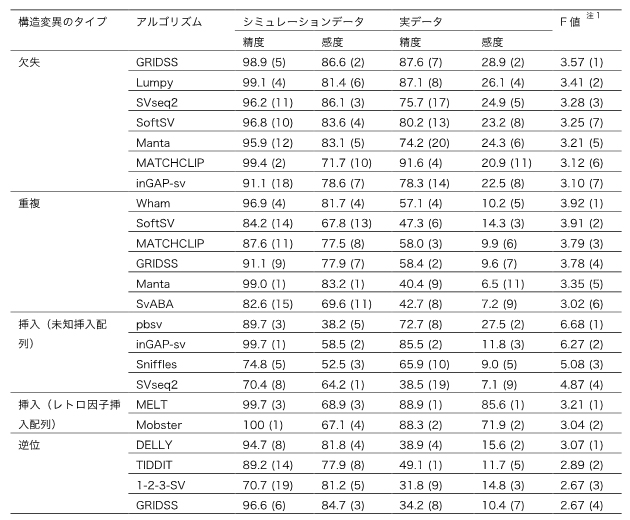
表1 各SVタイプで良い性能を示すアルゴリズムのリスト
注1は、シミュレーションデータで得られたF値と実データで得られたF値の和を示す。
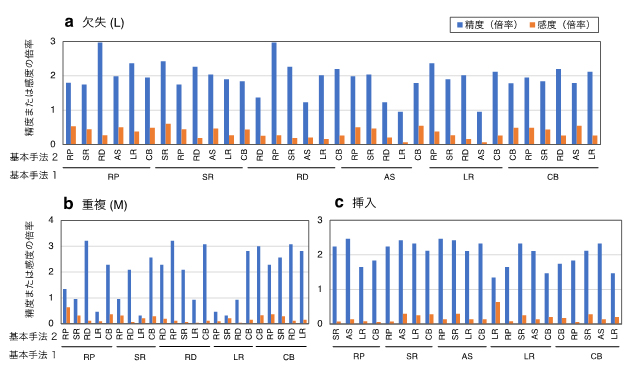
図3 SV検出アルゴリズム間で共通に検出されるSVの精度と感度
NA12878実データを用いて各アルゴリズムから検出された欠失(サイズ: L)(a)、重複(サイズ: M)(b)、挿入(c)の精度と感度が計測された。さらに、アルゴリズム間(総当たりペア)で共通に検出されるSVの精度と感度が計測された。単独アルゴリズムAの精度(および感度)に対するアルゴリズムペアA-B間で共通に検出されるSVの精度(および感度)の比が計算された。得られた結果は、アルゴリズムそれぞれに用いられている六つの基本手法(RP: read pair、SR: split read、RD: read depth、AS: assembly、LR: long read、CB: RP, SR, RD, ASを組み合わせた手法)に基づいて分類・集約された。示された結果は、基本手法1を持つ単独アルゴリズムの精度(および感度)に対する、基本手法1と基本手法2をそれぞれに持つアルゴリズムペア間で共通に検出されるSVの精度(および感度)を集約したものである。

