
2021-03-12 横浜市立大学,理化学研究所,日本医療研究開発機構
横浜市立大学大学院医学研究科遺伝学髙田篤客員准教授(理化学研究所脳神経科学研究センター分子精神病理研究チーム チームリーダー兼務)、濱中耕平助教、松本直通教授の研究グループは、遺伝統計学的解析と人工知能(機械学習)を用いたデータ駆動型アプローチ(注1)で、米国臨床遺伝・ゲノム学会(American College of Medical Genetics and Genomics:ACMG)と分子病理学会(Association for Molecular Pathology:AMP)が作成した世界標準の臨床遺伝子診断ガイドライン(ACMGガイドライン(注2))を洗練させるための手法を開発し、報告しました。これにより、日本発の技術で、世界標準たるガイドラインの「カイゼン」を達成しました。本研究の成果により、既存のガイドラインに従った判定では見逃されていた遺伝性疾患の原因遺伝子変異を発見したり、誤診を回避したりすることが可能になると予想されます。
本研究は、Cell出版社のトランスレーショナル医学雑誌『Med』に掲載されます。(日本時間2021年3月12日午前1時付オンライン)
研究成果のポイント
- 大規模ゲノム変異データの遺伝統計解析で、各変異タイプの平均的有害度を推測する方法を確立
- その方法を用いて各変異タイプを解析し、ACMGガイドラインでは、転写産物の翻訳開始コドンの変異(スタート喪失変異)と、転写産物の読み枠のずれが生じない挿入欠失変異(インフレーム挿入欠失変異(注3))の病原性が過大評価され、転写産物の翻訳終止コドンの変異(ストップ喪失変異)の病原性が過小評価されていることを明らかに
- 引き続いて、ストップ喪失変異とインフレーム挿入欠失変異のうち、病原性が高いと推測されるサブグループを、既存の生物学的知識や解析ツールを用いて抽出する方法を確立
- 既存の知識では有害性予測が困難であったスタート喪失変異について、その病原性を予測するモデルを機械学習で開発
- データ駆動型のアプローチで、知識ベースの診断ガイドラインを洗練させるための手法を提示
研究支援
本研究は、AMEDゲノム医療実現推進プラットフォーム事業(先端ゲノム研究開発)「オリゴジェニックモデルに基づくヒト疾患の遺伝的構造の解析」(代表・髙田篤)、難治性疾患実用化研究事業「新技術を用いた難治性疾患の高精度診断法の開発」(代表・松本直通)、「遺伝統計学的解析によるてんかん性脳症の新規原因遺伝子探索及び病態解明」(代表・髙田篤)、脳科学研究戦略推進プログラム「トリオサンプルのシーケンス解析による、遺伝子型によって定義される双極性障害の一群の同定」(代表・髙田篤)、日本学術振興会などによる支援を受けて行われました。
研究の背景
ヒト遺伝性疾患の分子診断(タンパク質やDNAなどの分子を調べて疾患を特定する)を正確に行うためには、遺伝子変異の病原性、変異の遺伝形式、臨床症状など、様々な情報を統合する必要があります。米国臨床遺伝・ゲノム学会(American College of Medical Genetics and Genomics:ACMG)と分子病理学会(Association for Molecular Pathology:AMP)によって、2015年にヒト遺伝性疾患の分子診断における診断ガイドライン「ACMGガイドライン」が作成され、世界標準として幅広く利用されています(論文検索システムであるGoogle Scholarでの引用回数は2021年3月時点で9700回以上)。
ACMGガイドラインでは、例えば、ある疾患の原因遺伝子上のDNA塩基配列(注4)の変異の候補が、遺伝子がコードするタンパク質の機能を完全に喪失させると予想される、ノンセンス変異(注5)やフレームシフト挿入欠失変異(注6)であった場合には、病原性を示唆するエビデンスとして最も強い「PVS(pathogenic very strong:以下、「超強力」と表現)」の基準を満たすと判定されます。同様に、患者でのみ認められ、両親では認められない突然変異であった場合には、強いエビデンスである「PS(pathogenic strong:以下、「強力」と表現)」の基準、変異が重篤な先天性疾患を認めないヒトの集団では観察されない場合には、中等度のエビデンスである「PM(pathogenic moderate:以下、「中等度」と表現)」の基準、変異が複数のコンピュータプログラムで有害と予測されるミスセンス変異(注7、以下「有害ミスセンス変異」と表現)である場合は、弱いが支持的なエビデンスである「PP(pathogenic supportive:以下、「支持的」と表現)」の基準を満たすと、それぞれ判定されます。ACGMガイドラインでは、このような基準の組み合わせから、変異の病原性を判定します。例えば、PVSの基準を満たし、かつ「中等度」の基準を2つ以上満たす場合や、「強力」の基準を1つ満たし、かつ「中等度」の基準を3つ以上満たす場合、その遺伝子変異は「病原性あり(pathogenic)」と判断されます。
このガイドラインの基準には、変異の機能的タイプ(例えば上述のノンセンス変異、フレームシフト変異など)に基づくものがいくつかあり、タンパク質の翻訳開始コドンが変化して、翻訳開始位置が変わるかなくなってしまうと予想される「スタート喪失変異」は「超強力」と判定されます。また、タンパク質の翻訳終止コドンが変化して、通常よりも長い異常なタンパク質が翻訳されると予想される「ストップ喪失変異」は、タンパク質の読み枠を変化させない「インフレーム挿入欠失変異」とともに「中等度」に分類されます。
一方、「スタート喪失変異」に関しては、例えば近傍に別の開始コドンが存在する場合には遺伝子機能に対する影響が大きくない可能性があります。また、多数の異常なアミノ酸が付与されることが多い「ストップ喪失変異」を、数個のアミノ酸の変化にしかつながらない「インフレーム挿入欠失変異」とひとくくりにして良いのかという問題もあります。さらに、このガイドラインはエキスパートの知識・意見にのみ依拠しており、客観的データに基づいて吟味されたものではありません。
研究グループはこれらの問題に対して、遺伝統計学、機械学習といった、近年注目を浴びているデータ駆動型のアプローチで取り組みました。
研究の内容
研究グループはまず、ヒト一般集団のデータを用いて、様々なタイプの変異の平均的な有害度を推測し、統計学的に評価するための方法を確立することを試みました。これまでに他グループが行った研究や、今回研究グループが行ったシミュレーション解析(図1左)の結果から、平均的有害度が高く、変異が次世代に引き継がれる確率が低いタイプの変異(例えば幼少期発症の重篤な疾患の原因変異など)ほど、変異の頻度分布が稀な変異(棒グラフ中の濃い赤の部分、頻度0.001%未満)に偏ることが示されています。研究グループはこれを、大規模一般集団ゲノムデータベースであるgnomAD(注8)の、12万人以上の遺伝子変異データを用いて検証しました。その結果、既存の各変異タイプの平均的有害度に対する知見と一致する形で、機能喪失変異(ノンセンス変異やフレームシフト挿入欠失変異など、スタート喪失変異を含まない)で最も稀な変異が多く、続いて有害ミスセンス変異、有害と予測されないミスセンス変異、タンパク質を変化させないシノニマス変異(注9)の順で、稀な変異が多いことが示されました(図1右上)。また、それぞれの変異タイプにおける稀な変異の割合の違いが偶然の範囲内なのか、統計学的に意味がある差なのかを、フィッシャーの正確検定(注10)という統計解析手法で評価しました。すると、各タイプ間で統計学的に有意な(=偶然では起こりえない)差があることが分かりました。
 図1 頻度分布に基づく変異タイプの有害性推定左パネル:頻度別の色分けと(上)と、変異をランダムに生成したシミュレーション解析における有害もしくは無害な変異の頻度分布。解析では、アフリカで誕生し世界中に広がっていった民族移動の歴史もシミュレートしている。右上パネル:gnomADの実世界データを用いた解析結果。一般的な変異の平均的有害度の理解(機能喪失>有害ミスセンス>その他ミスセンス>シノニマス)と一致した形で、稀な変異(濃い赤の頻度0.001%未満)の割合が異なることが示された(黒線は稀な変異の割合の95%信頼区間を示す)。右下パネル:gnomADの実世界データを用いて、スタート喪失変異、ストップ喪失変異、インフレーム挿入欠失変異について評価した結果。右側にACMGガイドラインでの判定を示す。この結果に基づくと、ACMGガイドラインではスタート喪失変異とインフレーム挿入欠失変異の病原性が過大評価され、ストップ喪失変異の病原性が過小評価されている可能性がある。
図1 頻度分布に基づく変異タイプの有害性推定左パネル:頻度別の色分けと(上)と、変異をランダムに生成したシミュレーション解析における有害もしくは無害な変異の頻度分布。解析では、アフリカで誕生し世界中に広がっていった民族移動の歴史もシミュレートしている。右上パネル:gnomADの実世界データを用いた解析結果。一般的な変異の平均的有害度の理解(機能喪失>有害ミスセンス>その他ミスセンス>シノニマス)と一致した形で、稀な変異(濃い赤の頻度0.001%未満)の割合が異なることが示された(黒線は稀な変異の割合の95%信頼区間を示す)。右下パネル:gnomADの実世界データを用いて、スタート喪失変異、ストップ喪失変異、インフレーム挿入欠失変異について評価した結果。右側にACMGガイドラインでの判定を示す。この結果に基づくと、ACMGガイドラインではスタート喪失変異とインフレーム挿入欠失変異の病原性が過大評価され、ストップ喪失変異の病原性が過小評価されている可能性がある。
引き続いて、機能喪失変異(ACMGガイドラインで「超強力」)、有害ミスセンス変異(「支持的」)とともに、スタート喪失変異、ストップ喪失変異、インフレーム挿入欠失変異における稀な変異の割合を評価しました。すると、スタート喪失変異(「超強力」)とインフレーム挿入欠失変異(「中等度」)の稀な変異の割合は有害ミスセンス変異(「支持的」)よりも小さく、ACMGガイドラインではスタート喪失変異とインフレーム挿入欠失変異の有害度が過大評価されていると考えられました。また、ストップ喪失変異(「中等度」と判定)の稀な変異の割合は機能喪失変異(「超強力」)に近く、その病原性が過小評価されていると考えられました。(図1右下)。
この結果は世界標準たるACMGガイドラインの精度に疑問を投げかけるものですが、ただ批判を行うだけではあまり建設的ではありません。そこで、研究グループは、スタート喪失変異、ストップ喪失変異、インフレーム挿入欠失変異の中で、より有害度が高いサブグループを抽出することを試みました。その結果、ストップ喪失変異については、変異によって付与される異常なアミノ酸の数が多いグループ(上位25%)で特に稀な変異が多く、機能喪失変異(ACMGガイドラインで「超強力」)と同程度の有害度を有すると推測されること(図2左上)が分かりました。また、付与される異常なアミノ酸の数と変異の頻度の間には、有意な相関が認められました(図2右上)。インフレーム挿入欠失変異については、その有害度を予測する既存のコンピュータプログラムがいくつかあります。研究グループは複数のプログラムを用いて検討し、PROVEAN(注11)というプログラムで有害と予測される変異には稀な変異が多く、有害ミスセンス変異(ACMGガイドラインで「支持的」)と同程度であること(図2左下)、PROVEANの有害度スコアと変異の頻度が有意に相関すること(図2右下)が分かりました。
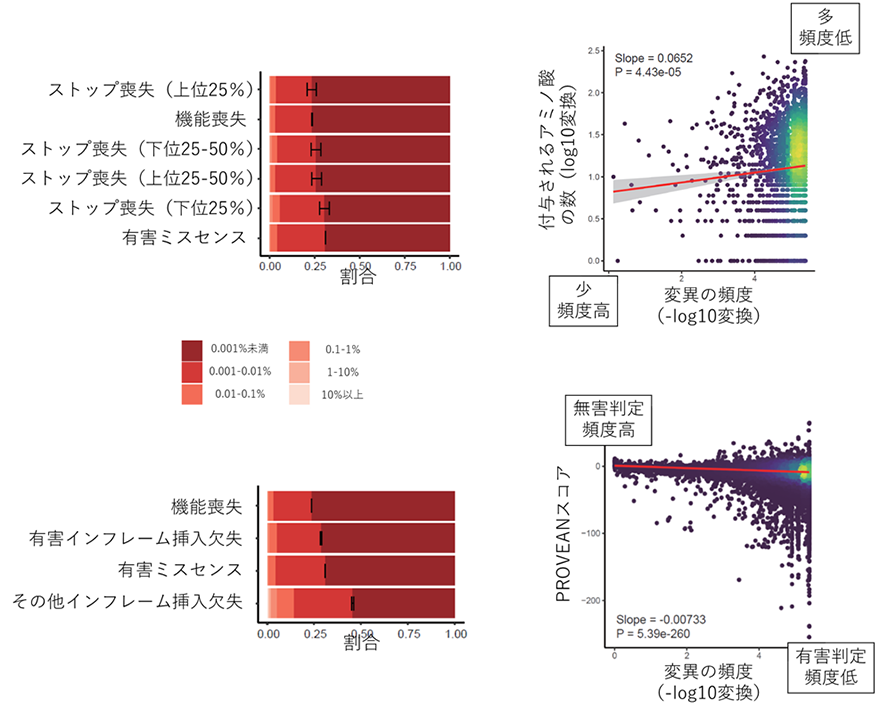 図2 稀な変異が多いストップ喪失変異とインフレーム挿入欠失変異のサブグループ左上パネル:ストップ喪失変異を、付与される異常なアミノ酸の数が多い順で分けた4つのグループと、機能喪失変異、有害ミスセンスの比較。付与されるアミノ酸の多さでトップ25%(32アミノ酸以上)のストップ喪失変異グループは、機能喪失変異と同程度の稀な変異の割合を示した。黒線は稀な変異の割合の95%信頼区間を示す。右上パネル:各ストップ喪失変異の頻度と付与される異常なアミノ酸の数の散布図。各点が各変異を示す。点の色は点の密度を示す(黄色が高密度のエリア)。赤線は回帰直線、灰色エリアは回帰直線の信頼区間を示す。付与されるアミノ酸が多く頻度が低い変異は図の右上、その反対は左下にプロットされる。回帰直線が右肩上がりとなっており、付与されるアミノ酸が多いほど頻度が低くなる傾向が示されている。左下パネル:PROVEANで有害と判定されるインフレーム挿入欠失変異、その他のインフレーム挿入欠失変異、機能喪失変異、有害ミスセンス変異の比較。有害インフレーム挿入欠失変異は害ミスセンス変異と同程度の稀な変異の割合を示す。右下パネル:各インフレーム挿入欠失変異の頻度とPROVEANスコア(値が小さいほど有害)の散布図。有害判定で頻度が低い変異は図の右下、その反対は左上にプロットされる。点と線の色の意味は右下パネルと同じ。回帰直線は右肩下がりとなっている。
図2 稀な変異が多いストップ喪失変異とインフレーム挿入欠失変異のサブグループ左上パネル:ストップ喪失変異を、付与される異常なアミノ酸の数が多い順で分けた4つのグループと、機能喪失変異、有害ミスセンスの比較。付与されるアミノ酸の多さでトップ25%(32アミノ酸以上)のストップ喪失変異グループは、機能喪失変異と同程度の稀な変異の割合を示した。黒線は稀な変異の割合の95%信頼区間を示す。右上パネル:各ストップ喪失変異の頻度と付与される異常なアミノ酸の数の散布図。各点が各変異を示す。点の色は点の密度を示す(黄色が高密度のエリア)。赤線は回帰直線、灰色エリアは回帰直線の信頼区間を示す。付与されるアミノ酸が多く頻度が低い変異は図の右上、その反対は左下にプロットされる。回帰直線が右肩上がりとなっており、付与されるアミノ酸が多いほど頻度が低くなる傾向が示されている。左下パネル:PROVEANで有害と判定されるインフレーム挿入欠失変異、その他のインフレーム挿入欠失変異、機能喪失変異、有害ミスセンス変異の比較。有害インフレーム挿入欠失変異は害ミスセンス変異と同程度の稀な変異の割合を示す。右下パネル:各インフレーム挿入欠失変異の頻度とPROVEANスコア(値が小さいほど有害)の散布図。有害判定で頻度が低い変異は図の右下、その反対は左上にプロットされる。点と線の色の意味は右下パネルと同じ。回帰直線は右肩下がりとなっている。
さらに研究グループは、多数の異常なアミノ酸の付与を引き起こすストップ喪失変異、PROVEANで有害と判定されるインフレーム挿入欠失変異が、実際に病気の原因として報告されている変異に多いことを、ヒト疾患の原因・関連遺伝子変異の情報を収集した別のデータベースであるHGMD(注12)、ClinVar(注13)を用いて実証しました。
一方、スタート喪失変異については、変異のすぐそばに別の開始コドンが存在するかどうかなどの既存知識に基づく単純なルールだけではうまく有害度が高い変異を抽出することができませんでした。そこで研究グループは、近年注目されている人工知能(機械学習)技術を用いて、この問題の解決を図りました。まず、機械学習の一手法である深層学習(注14)を用いて開発された、転写産物中の翻訳開始点になりうる塩基配列とその翻訳開始点としての強さを予測するモデル(他グループが開発したTITER(注15)という手法)を用いて、各遺伝子の潜在的な翻訳開始点についての情報を集約しました。引き続いて、この情報と、既知翻訳開始点と潜在的翻訳開始点の距離などの情報を統合し、HGMD、ClinVarの既知疾患原因スタート喪失変異と、ヒト一般集団で認められ病気とは関係がないと考えられる変異を判別するモデルを、ランダムフォレスト(注16)という別の機械学習手法を用いて構築し、これをPoStaL(注17)と名付けました。PoStaLと、既存のスタート喪失変異にも使えるが特化はされていない変異の病原性予測プログラム(PolyPhen2(注18)など)との性能比較を行ったところ、PoStaLは圧倒的な優位性を示しました(図3)。この結果は、特定の変異タイプに最適化されたモデルを構築することの重要性を示しています。最後に研究グループは、PoStaLで特異度95%をもって病原性が高いと予測される変異は、ACMGガイドラインで「支持的」と判定される、有害ミスセンス変異と同程度に稀な変異が一般集団で多い(≒同程度の病原性を示す)ことを示しました。
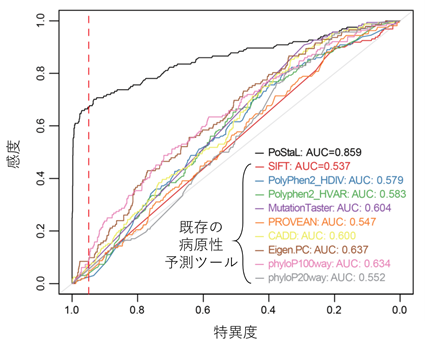
図3 既知疾患原因スタート喪失変異の判別性能各ツールの性能をROC曲線(注19)で評価したところ、PoStaL(黒線)が断トツで大きいarea under the curve(AUC:カーブの下の面積のことで判別ツールの総合的性能の指標の一つ。大きいほど性能が高いことを意味する)を示した。赤の点線は特異度95%のライン。黒線以外は、スタート喪失変異に特化はしていない(主にミスセンス変異などを対象とした)病原性予測ツールを用いたときの結果。
今後の展開
本研究の成果を、グラフィカルアブストラクト(図解した要旨)としてまとめると図4のようになります。この結果を参照しながら、さらに議論を深めることで、世界標準の遺伝子診断であるACMGガイドラインの精度をより向上させることができると予想されます。一方、臨床遺伝子診断においては、ACMGガイドラインのような基準は非常に有用ではあるものの、臨床症状を含めて個々のケースを詳細に評価することが極めて重要です。今回の研究は、臨床症状についても考慮した枠組みを提供するものではありませんが、そういった情報を統合していくことで、より洗練されたガイドラインを構築することができると予想されます。
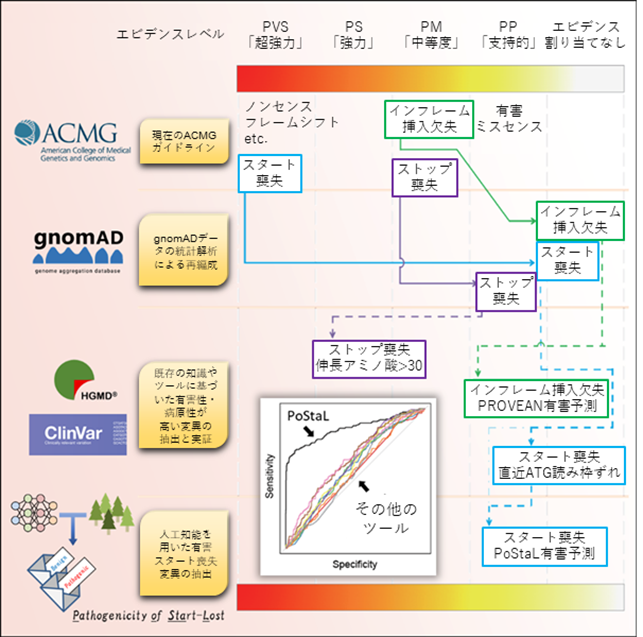
図4 本研究のグラフィカルアブストラクト現在のACMGガイドラインにおけるスタート喪失変異(PVS:「超強力」)、ストップ喪失変異(PM:「中等度」)、インフレーム挿入欠失変異(PM:「中等度」)の判定を1段目に示す。2段目は、gnomADデータを用いたこれらの変異の判定の再編成を、3段目は、既存の知識やツールを用いた、これらの変異の中の有害なサブグループの抽出法とHGMD、ClinVarデータを用いた検証をそれぞれ示す。一番下の4段目は、人工知能を用いた有害スタート喪失変異の判別モデル(PoStaL)の開発と評価を示す。
また本研究成果は、直接的に遺伝子診断ガイドラインの精度向上に繋がるだけではなく、データ駆動型のアプローチで、知識ベースのシステムをカイゼンするための方法の具体的な一例を提示するものです。さまざまな分野で利用されている、専門家によって集約された知識や意見を主たる根拠とした診断フレームワークやガイドライン、例えば精神科領域におけるDSM(注20)などを、今後データ科学でより良いものとしていく上でも、本研究の方法と成果はヒントにもなるかもしれません。
用語説明
- (注1)データ駆動型アプローチ
- ひとつの計算によって生成されるデータが次の計算を起動し、次々に一連の計算が実行されるように、使用可能なデータからスタートし、推論規則を使って最適解に達するまでさらにデータを引き出していく前向き連鎖の手法
- (注2)ACMGガイドライン
- 米国臨床遺伝・ゲノム学会(American College of Medical Genetics and Genomics:ACMG)と分子病理学会(Association for Molecular Pathology:AMP)が共同で発表した、遺伝子診断のガイドライン。下記論文に詳述されている。
Richards et al., Genetics in Medicine volume 17, pages405–423(2015), doi: 10.1038/gim.2015.30 - (注3)インフレーム挿入欠失変異
- 3で割り切れる数のDNA塩基配列の欠失もしくは挿入が認められるタイプの遺伝子変異。変異以降のタンパク質読み枠は維持される。
- (注4)DNA塩基配列
- ゲノムを構成するDNA塩基配列は、グアニン(G)、シトシン(C)、アデニン(A)、チミン(T)の4種類からなる。
- (注5)ノンセンス変異
- タンパク質翻訳を終了させる終始コドン(TAG、TAA、TGAのいずれか)が遺伝子の途中で生成されるタイプの遺伝子変異。
- (注6)フレームシフト挿入欠失変異
- 3で割り切れない数のDNA塩基配列の欠失もしくは挿入が認められるタイプの遺伝子変異。3つの塩基(コドン)で一つのアミノ酸がコードされるため、3で割り切れない数の挿入欠失が生じると、タンパク質の読み枠(フレーム)がずれて、それ以降のタンパク質配列が通常と全く異なるものとなってしまう。
- (注7)ミスセンス変異
- タンパク質中のアミノ酸を一つだけ通常と異なるものに変化させるタイプの遺伝子変異。
- (注8)gnomAD
- Genome Aggregation Databaseの略。米国ブロード研究所が作成したヒト遺伝子変異の大規模データベース。本研究で使用したバージョン2では一般集団125,748人のタンパク質コード領域の変異データと、15,708人の全ゲノム領域の変異データがまとめられている。
URLは https://gnomad.broadinstitute.org/ 。 - (注9)シノニマス変異
- DNA配列は変化するが、タンパク質中のアミノ酸は変化させないタイプの遺伝子変異。例えばCAAというコドンとCAGというコドンはいずれもアミノ酸はグルタミンをコードするので、このコドンの3番目のAがGに変化してもアミノ酸は変化しない。
- (注10)フィッシャーの正確検定
- 2つ以上のカテゴリーに分類されたデータの分析に用いられる統計学的検定法。英国の統計学者ロナルド・フィッシャーが考案した。
- (注11)PROVEAN
- Protein Variation Effect Analyzerの略。J.クレイグベンター研究所のグループが開発した遺伝子変異の有害性予測ツール。
URLは http://provean.jcvi.org/index.php 。 - (注12)HGMD
- Human Gene Mutation Databaseの略。英国カーディフ大学が運営する、既知のヒト疾患原因・関連変異のデータベース。
- (注13)ClinVar
- 米国National Center for Biotechnology Information(NCBI)が提供する、ヒト疾患原因・関連変異のデータベース。
- (注14)深層学習
- 多層のニューラルネットワーク(ディープニューラルネットワーク)による機械学習の手法。ディープラーニングともよばれる。
- (注15)TITER
- Translation Initiation siTE detectoRの略。転写産物中の関心部位の周辺配列をもとに、その部位がタンパク質翻訳開始点となりうるかどうかを深層学習で評価するソフトウェア。中国清華大学のグループが開発。
URLは https://github.com/zhangsaithu/titer 。 - (注16)ランダムフォレスト
- ランダムにサンプリングしたデータと説明変数を用いて、多数の決定木(決定を行うための分岐のグラフ)を作り、各決定木の予測結果を統合してモデルを構築する機械学習アルゴリズム。
- (注17)PoStaL
- Pathogenicity of Start-Lostの略。本研究で開発した、スタート喪失変異の病原性予測モデル。Postalは英語で「郵便の」の意。スタート喪失変異が病原性を有するかどうかを知らせるお手紙が届くイメージ。
- (注18)PolyPhen2
- Polymorphism Phenotyping v2の略。ハーバード大学のグループが開発した遺伝子変異の有害性予測ツール。
URLは http://genetics.bwh.harvard.edu/pph2/ 。 - (注19)ROC曲線
- 特異度と感度の変化を二次元平面上にプロットして分類予測の性能を表した曲線。ROCはReceiver Operatorating Characteristicの略。もともとはレーダー信号のノイズの中から敵機の存在を検出するための方法として開発された。
- (注20)DSM
- Diagnostic and Statistical Manual of Mental Disordersの略。精神疾患の分類のための共通言語と標準的な基準を提示するフレームワーク。何度か改訂がなされ、現在はDSM-5が使用されている。
掲載論文
- タイトル
- Refinement of clinical variant interpretation framework by statistical evidence and machine learning
- 著者
- Atsushi Takata, Kohei Hamanaka, Naomichi Matsumoto
- 掲載誌
- Med 2, 1-22, April 9 (2021)
- URL
- https://www.cell.com/med/fulltext/S2666-6340(21)00064-7
お問い合わせ先
本資料の内容に関するお問い合わせ
横浜市立大学大学院医学研究科 遺伝学 客員准教授
理化学研究所 脳神経科学研究センター
分子精神病理研究チーム チームリーダー
髙田篤
横浜市立大学大学院医学研究科 遺伝学 教授
松本直通
取材対応窓口、詳細の資料請求など
横浜市立大学 広報室 担当課長 上村一太郎
理化学研究所 広報室 報道担当
AMEDの事業について
国立研究開発法人日本医療研究開発機構
ゲノム・データ基盤事業部 ゲノム医療基盤研究開発課

