
2021-04-09 浜松医科大学,横浜市立大学,日本医療研究開発機構
研究成果のポイント
- てんかんと発達の遅れがある小児患者700例のゲノムDNAの配列を次世代シークエンスで決定することで、新たな責任遺伝子ATP6V0A1の異常を発見しました
- ATP6V0A1遺伝子は、リソソーム(タンパク質の分解に関わる細胞内小器官)の酸性度を調節するプロトンポンプを産生します。
- 患者と同じ遺伝子異常を導入したマウスモデルを詳細に解析することで、遺伝子異常がリソソームの機能異常をもたらし、神経と神経のつなぎ目であるシナプス形成を障害することを明らかにしました。
- 今後、このモデルマウスを用いた研究によって効果的な治療法の開発が進むことが期待されます。
※本研究成果は、英国科学雑誌「Nature Communications」に日本時間2021年4月8日(木)午後7時に公表されます。
概要
浜松医科大学医化学の青戸一司助教・才津浩智教授、横浜市立大学遺伝学の松本直通教授、浜松医科大学神経生理学の秋田天平准教授、福田敦夫教授、昭和大学小児科学の加藤光広教授らの共同研究グループは、発達性およびてんかん性脳症(developmental and epileptic encephalopathy: DEE)の新たな責任遺伝子(異常があると当該の病気が発症する遺伝子)ATP6V0A1を発見しました。この遺伝子が産生するタンパク質ATP6V0A1は、タンパク質の分解に関わる細胞内小器官であるリソソームの酸性度を調節するプロトン(H+)ポンプの構成サブユニットの1つで、1) リソソームを酸性に保つことで分解酵素の働きを維持する役割、2) 細胞の増殖を促進するmTORシグナルを調節する役割、3) 細胞内のタンパク質等を分解する仕組みであるオートファジーにおいて、分解予定のタンパク質を取り込んだオートファゴゾームとリソソームを癒合させる役割が知られていました(図1左)。
本研究では、ATP6V0A1変異を4名のDEE症例で同定しました。また、症例と同様の変異を持ったAtp6v0a1変異マウスの解析により、上記のATP6V0A1の機能が障害されることに加えて、神経と神経のつなぎ目であるシナプスの数が減少することを明らかにしました(図1右)。このマウスモデルを用いた研究により、効果的な治療法の開発への寄与が期待されます。

図1. ATP6V0A1の機能と疾患発症のメカニズム
オレンジ色の〇がリソソーム、青い〇がオートファゴゾームを表す。
研究の背景
てんかんは最も頻度が高い神経疾患の一つで、日本国内に人口の1%近くの約100万人の患者がいると推定されています。てんかんは外傷、感染症、脳出血、脳腫瘍など様々な原因が知られていますが、最も頻度の高い原因は遺伝子の異常によるものであるといわれています。特に早期発症型てんかんにおいては遺伝要因の関与が強く示唆されていますが、関与する遺伝子異常は多彩であり、近年の次世代シークエンス技術の発展によって多数の責任遺伝子の異常が明らかになってきています。
また、2020年にノーベル化学賞を受賞した遺伝子を書き換えることのできるゲノム編集技術の登場により、患者と同じ遺伝子変異を持ったモデルマウスの作製が容易になりました。モデルマウスが患者の症状を模倣している場合には、その解析を通じて病気の発症機序の解明が進むことが期待されます。
研究の成果
1. DEE患者におけるATP6V0A1遺伝子変異の発見
研究グループは、DEEの原因遺伝子を探るために700例のDEE患者からDNAを採取し、次世代シークエンサーを用いた全エクソーム解析*1を行いました。その結果、2名の患者(患者1、2)において、ATP6V0A1遺伝子の同一の突然変異(p.R741Q、741番目アミノ酸のアルギニンがグルタミンに置換)を同定し、更に別の2名の患者(患者3、4)において、ATP6V0A1遺伝子の両アレル性変異*2を同定しました【そのうち患者3が遺伝子欠失とA512P変異(512番目のアラニンがプロリンに置換)、患者4がスプライス部位の変異とN534D変異(534番目のアスパラギンがアスパラギン酸に置換)】。全ての患者で、知的障害、発達遅滞、てんかんと脳萎縮を認めており、特に患者3においては進行する重度の脳萎縮を認めました(図2)。
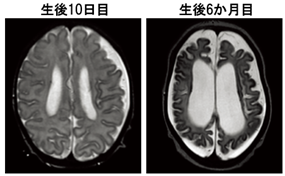
図2. 患者3の生後10日目と生後6か月目の脳MRI所見生後10日目では軽度の脳萎縮が認められるが、生後6か月では重度の脳萎縮が認められ、進行性であることが分かる。
2. A512P変異、N534D変異、R741Q変異はATP6V0A1の機能を障害する
変異がATP6V0A1タンパク質の機能に与える影響を調べるために、3つの変異遺伝子(A512P変異、N534D変異、R741Q変異)を発現させた培養細胞におけるリソソームの酸性度を調べたところ、全ての変異においてプロトンポンプ機能の異常を示唆する酸性度の異常が観察されました(図3)。さらに、CRISPR-Cas9ゲノム編集技術*3を用いて、3つの変異のうちR741Q変異とA512P変異を導入して変異マウスをそれぞれ作製したところ、R741Q変異のホモ接合性マウス(*2の説明文参照)は母獣の胎内で死亡するのに対して、A512P変異のホモ接合性マウスは生まれるものの、生後すぐに体重増加の不良や、立ち直りがうまくできないといった運動失調がみられ、2週間以内に死亡しました。このことから、R741Q変異、A512P変異ともにATP6V0A1の機能を障害すること、R741Q変異の方がより重度に機能を障害することが明らかとなりました。
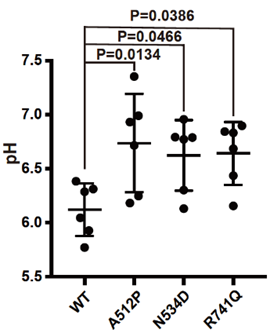
図3. DEE患者で同定された3つの変異を過剰に発現させた培養細胞でのリソソームの酸性度(pH)3つの変異体を発現させた細胞では野生型(WT)を発現させた細胞と比較して酸性度が増加しており、プロトンポンプ機能が障害されていると考えられる。
3. A512P変異ホモ接合性マウスでは神経のつなぎ目であるシナプスの数が減少する
A512P変異ホモ接合性マウスが示す異常を詳細に解析することは、ATP6V0A1変異が原因となるDEEの発症機序を明らかにすることに繋がります。A512P変異ホモ接合性マウスの大脳皮質、海馬、小脳といった脳の各部位では、神経細胞の減少に加えて、活性を持ったリソソーム酵素の減少、mTORシグナルの減少が認められました。これらの所見は、ATP6V0A1の機能が変異マウスの脳で障害されていることを示しており、患者で認められた脳の萎縮を反映していると考えられました。また、電子顕微鏡で生後10日目の神経細胞を詳しく観察したところ、細胞内の老廃物や不要物を取り込んだオートファゴゾームとリソソームとの癒合が障害されて、それらが細胞内に蓄積している様子が観察されました(図4A)。更に、神経と神経のつなぎ目であるシナプスの数が海馬や小脳で減少していることが分かり、ATP6V0A1がシナプス形成に重要な役割を果たしていることが明らかになりました(図4B)。
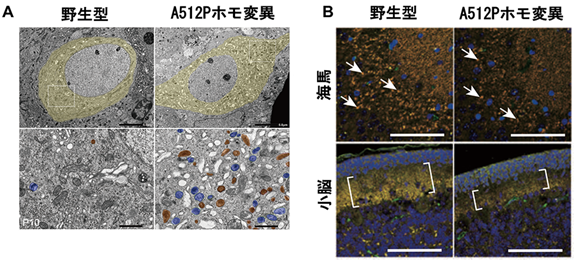
図4. A512P変異マウス脳の異常所見(A)海馬神経細胞の電子顕微鏡画像。青がオートファゴソーム、茶色がリソソームを示す。A512Pホモ接合性変異マウスではオートファゴソームとリソソームが神経細胞内で著明に増加している。
(B)海馬および小脳におけるシナプスの数。前シナプスのマーカー(赤)と後シナプスのマーカー(緑)がともに染色されるシナプス(黄色)の数が、A512Pホモ接合性変異マウスでは半分以下に減少している(上段では一部を矢印で示す、下段ではカッコで示される領域)。
注釈
- *1全エクソーム解析:
- ゲノム上の全エクソン領域(タンパク質の配列を決定する遺伝子領域の全て)を分離した後、その塩基配列を次世代シークエンサーで決定する方法。
- *2 両アレル性変異:
- 常染色体において、父親から受け継いだ染色体と母親から受け継いだ染色体の両方のアリル(遺伝子)に変異があること。2つのアリル(遺伝子)の変異が同一の場合と異なる場合があり、同一の場合はホモ接合性変異と呼ぶ。
- *3 CRISPR-Cas9ゲノム編集:
- 2020年にノーベル化学賞を受賞した生物の狙った遺伝子を書き換えたり、削除したりすることのできる遺伝子改変技術。
今後の展開
本研究は、リソソームの膜タンパク質ATP6V0A1の変異がDEEに関与していることを明らかにし、疾患モデルマウスを解析することで、変異がリソソームの機能異常をもたらし、シナプス形成を障害することを明らかにしました。今後、このモデルマウスを用いた研究によって効果的な治療法の開発が進むことが期待されます。
論文情報
- 発表雑誌
- Nature Communications
- 論文タイトル
- ATP6V0A1 encoding the a1-subunit of the V0 domain of vacuolar H+-ATPases is essential for brain development in humans and mice
- 著者
- Kazushi Aoto, Mitsuhiro Kato, Tenpei Akita, Mitsuko Nakashima, Hiroki Mutoh, Noriyuki Akasaka, Jun Tohyama, Yoshiko Nomura, Kyoko Hoshino, Yasuhiko Ago, Ryuta Tanaka, Orna Epstein, Revital Ben-Haim, Eli Heyman, Takehiro Miyazaki, Hazrat Belal, Shuji Takabayashi, Chihiro Ohba, Atsushi Takata, Takeshi Mizuguchi, Satoko Miyatake, Noriko Miyake, Atsuo Fukuda, Naomichi Matsumoto, and Hirotomo Saitsu
研究グループ
本研究は、浜松医科大学医化学講座、横浜市立大学遺伝学講座、昭和大学小児科学講座、浜松医科大学神経生理学講座、新潟県はまぐみ小児療育センター・小児科・赤坂紀幸医師、国立病院機構西新潟中央病院・神経小児科・遠山潤医師、野村芳子小児神経クリニック・野村芳子医師、瀬川記念小児神経学クリニック・星野恭子医師、岐阜大学大学院医学系研究科小児病態学・吾郷耕彦医師、茨城県立こども病院・田中竜太医師、イスラエル・シャミール医療センター・小児神経発達センター・オルナ・エプスタイン医師、リバイタル・ベンハイム医師、エリ・ヘイマン医師、浜松医科大学医用動物資源支援部・高林秀次准教授との共同研究で、下記の助成により実施した成果です。
- 日本医療研究開発機構 難治性疾患実用化研究事業
- 2017~2019年度「ゲノム編集技術を用いた希少難治性神経発達障害の原因遺伝子変異ノックインマウスモデルの確立およびその解析による病態解明と新規治療薬探索」(研究開発代表者 才津浩智)
- 2017~2019年度「希少難病の高精度診断と病態解明のためのオミックス拠点の構築」(研究開発代表者 松本直通)
- 2018~2020年度「未診断疾患イニシアチブ(Initiative on Rare and Undiagnosed Diseases(IRUD)):希少未診断疾患に対する診断プログラムの開発に関する研究」(研究開発分担者 松本直通※)
※IRUDにおける解析センターとして参画。 - 2020年度~「新技術を用いた難治性疾患の高精度診断法の開発」(研究開発代表者 松本直通)
- 日本学術振興会科学研究費補助金 基盤研究(B)
- 「多面的アプローチによる小児脳神経疾患の遺伝要因と分子病態の解明」:研究代表者 才津浩智
- その他の日本学術振興会科学研究費、厚生労働省科学研究費補助金、武田科学振興財団研究助成金
お問い合わせ先
本件に関するお問い合わせ先
国立大学法人浜松医科大学 医化学講座
教授 才津 浩智
公立大学法人横浜市立大学 学術院医学群 遺伝学
教授 松本 直通
AMED事業について
国立研究開発法人日本医療研究開発機構
創薬事業部 創薬企画・評価課
難治性疾患実用化研究事業 担当

