
新型コロナウイルス感染症(COVID-19)の治療薬設計に役立つウイルスタンパク質と治療薬候補化合物の相互作用データを公開
2020-04-17 理化学研究所,星薬科大学,日本医療研究開発機構
理化学研究所(理研)生命機能科学研究センター制御分子設計研究チームの本間光貴チームリーダー、星薬科大学薬学部の福澤薫准教授らの共同研究グループは、新型コロナウイルス(SARS-CoV-2)タンパク質と治療薬候補化合物[1]の分子間相互作用を「フラグメント分子軌道法(FMO法)[2]」で計算し、データを、世界中の創薬研究者が自由に利用できる「FMOデータベース(FMODB)」注1)にて、4月17日に公開しました。
COVID-19の原因ウイルスであるSARS-CoV-2のゲノムには、ウイルス粒子を形作るタンパク質と、ウイルスゲノムの複製やウイルスタンパク質の成熟に関わる酵素などがコードされています。これらのタンパク質を標的とした治療薬開発のための基礎データとして、日本蛋白質構造データバンク(PDBj)のCOVID-19特集ページには、SARS-CoV-2が持つ約20種のウイルスタンパク質のうち9種について、ウイルスタンパク質単独あるいは治療薬候補化合物やヒト由来タンパク質との複合体の結晶構造[3](静的な立体構造)が131個登録されています注2)。これらの構造データは治療薬開発に非常に有用な情報ですが、次の段階として、治療薬候補化合物がウイルスタンパク質とどのような相互作用を形成するときに強く結合するかを精密に計算して明らかにすることが重要となります。
FMO法は、量子化学(第一原理)計算[2]により分子間相互作用を記述する手法です。今回共同研究グループは、PDBjに登録された131個の立体構造のうち、65個についてFMO計算を行い、治療薬候補化合物との相互作用(分子間にかかる引力、斥力などの力)を解析しました。図1に9種のタンパク質の構造を、図2にCH/π軌道相互作用が重要な役割を果たしている治療薬候補化合物の拡大図を示します。これらのデータは、タンパク質量子化学計算値データベースであるFMODBで公開され、ユーザーが、治療薬候補化合物がどのようなメカニズムで標的タンパク質と結合するかを精密に評価し、より強く標的に結合する効果の高い治療薬を設計する際に参考になります。
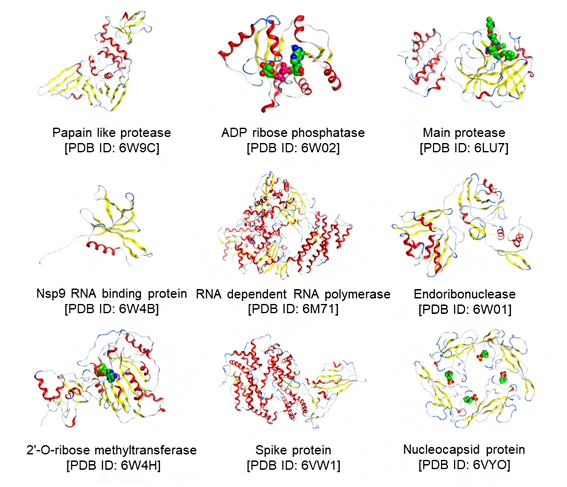
図1 FMO計算を実施した9種のタンパク質の代表構造
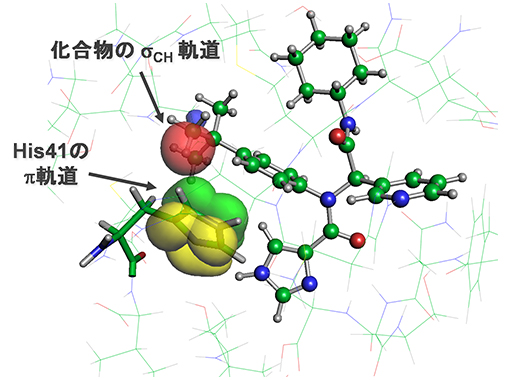
図2 メインプロテアーゼと治療薬候補化合物がCH/π軌道相互作用を形成している様子
ヒスチジン残基のπ軌道(黄色と緑色)が、化合物のσCH 軌道(赤色)と接近して引力相互作用を形成する。
注1)FMOデータベース(FMODB)は、BINDSの支援を受けて本間、福澤らがFMO創薬コンソーシアムとともに開発した世界初のタンパク質の量子化学計算値データベース。2019年3月公開。
注2)4月15日時点。最新情報は、大阪大学蛋白質研究所の日本蛋白質構造データバンク(Protein Data Bank Japan: PDBj)【COVID-19特集ページ】を参照。
背景
中国武漢に端を発した新型コロナウイルス(SARS-CoV-2)による感染症(COVID-19)は、2020年3月に入り、ヨーロッパ、北米が中心となる世界的な大流行(パンデミック)に至りました。COVID-19が世界の健康および経済活動に与える影響は甚大であり、多くの貴重な人命が失われ、都市封鎖(ロックダウン)に伴う社会基盤の損失は、かつてないレベルに達しつつあります。人類のほとんどがこの新興感染症に有効な免疫を持っていないことから、今後、感染者は数度の波を経て世界人口のかなりの割合に及ぶ可能性があり、有効な治療薬がないことが社会不安の原因になっています。
このような状況のもと、治療薬の開発は世界的に最も重要な課題となっており、世界の多くのグループが研究を行っています。治療薬の標的候補であるウイルスタンパク質には、ウイルス粒子を形作るスパイクタンパク質[4]、ヌクレオカプシドタンパク質[4]、ヒトの細胞に感染した後、細胞内で働く酵素となるメインプロテアーゼ[5]、RNA依存性ポリメラーゼ[5]などがあります。それらのタンパク質は、SARS-CoV-2では約20種が知られていますが、そのうち9種の結晶構造が既に解明されています。
日本蛋白質構造データバンク(PDBj)は早くから特集ページを作成して、これらのタンパク質構造情報を整理し、さまざまな条件や治療薬候補化合物などとの複合体について4月15日の時点で131個の構造を公開しています。これらの構造データは治療薬開発に非常に有用な情報ですが、次の段階として、治療薬候補化合物がウイルスタンパク質とどのような相互作用を形成するときに強く結合するかを精密に計算して明らかにすることが重要です。
「フラグメント分子軌道(FMO)法」は、量子化学(量子力学)計算により分子間相互作用を記述する手法であり、古典力学計算による分子動力学(MD)法[6]とともに、立体構造に基づいたインシリコ創薬[7]への応用が期待されます。SARS-CoV-2に対しては、立教大学理学部の望月祐志教授の研究チームが、メインプロテアーゼとその阻害剤候補化合物のFMO計算結果を3月に報告しました注3)。今回共同研究グループは、SARS-CoV-2関連タンパク質の構造全般にわたってFMO計算を実施し、ウイルスタンパク質と治療薬候補化合物の分子間相互作用データを公開することで、世界的な治療薬研究やワクチン開発の加速を目指しました。
研究手法と成果
共同研究グループは、ウイルスタンパク質と治療薬候補化合物との相互作用を解明するために、PDBjで公開された131個の結晶構造のうち65個に対して量子化学計算手法であるFMO法を適用し、精密な相互作用エネルギー値を明らかにしました。計算には理研のHOKUSAI(課題番号Q20306)、東京工業大学のTSUBAME3.0および最先端共同HPC基盤施設のOakforest-PACS(HPCIシステム産業利用課題hp200101)を用い、量子化学計算用ソフトウェアABINIT-MP[8]を使用しました。
計算された65個の創薬標的タンパク質における相互作用データは、全て「FMOデータベース(FMODB)」を通じて公開されており、トップページの本研究成果の特集ページから閲覧できます。一例として、代表的な標的タンパク質-治療薬候補化合物の複合体である、メインプロテアーゼ-治療薬候補化合物のFMOデータ(相互作用データ)の詳細閲覧画面を図3に示します。図3の上側が、構造データと計算の条件を示し、下のグラフは、治療薬候補化合物近傍のアミノ酸残基との相互作用についてエネルギー種別に表示しています。表示する範囲など、条件を変化させて見ることができます。
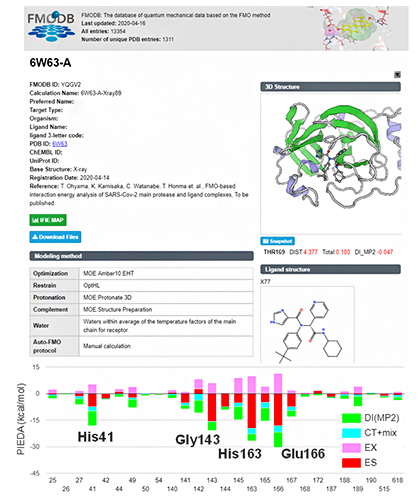
図3 FMOデータベース(FMODB)におけるメインプロテアーゼと治療薬候補化合物の相互作用データの一例
今後の期待
65個の創薬標的タンパク質のFMO法による相互作用データは、治療薬のどのような構造上の特徴がタンパク質と強く相互作用するかを解明し、世界的な治療薬設計の試みを加速すると期待できます。
理研の創薬・医療技術基盤プログラムでは、COVID-19特別プロジェクト注4)を設置して治療薬の研究開発を行っており、本研究は、理研の泰地真弘人らの研究チームによるタンパク質と治療薬候補化合物の動的な挙動を解明するための分子動力学シミュレーション注5)と並んで、研究の一翼を担っていきます。
注4)2020年3月30日お知らせ「COVID-19 特別プロジェクトの開始について」
注5)2020年3月23日プレスリリース「新型コロナウイルス(SARS-CoV-2)メインプロテアーゼの分子動力学シミュレーションデータを公開」
補足説明
1.治療薬候補化合物
創薬標的となるタンパク質と結合し、タンパク質の機能を変化させることで治療薬となる可能性のある化合物。多くの場合は、タンパク質の機能を効率よく阻害する働きを持つことが期待される。SARS-CoV-2に対しては、既存の抗ウイルス薬などからいくつかの化合物が治療薬候補として挙げられている。
2.フラグメント分子軌道法(FMO法)、量子化学(第一原理)計算
FMO法は、1999年に北浦和夫教授(当時大阪府立大学、現理研計算科学研究センター客員主管研究員)らによって開発された日本発の理論手法。タンパク質のような大きな分子系の物性を、量子力学の基本方程式(シュレーディンガー方程式)から求める量子化学(第一原理)計算を、アミノ酸残基や医薬品などの大きさのフラグメントに分割することによって可能とする。FMO法の利点として、フラグメント間の、すなわちアミノ酸残基と治療薬候補化合物の相互作用エネルギー(IFIE)を、静電相互作用や分散力相互作用など4種の異なる性質のエネルギー(PIEDA)に分けて算出することができる。FMOはFragment Molecular Orbitalの略。
3.結晶構造
X線結晶解析で得られた立体構造。タンパク質の結晶を作製し、その結晶にX線を照射して得られる回折データを解析することにより、タンパク質の内部の原子の立体的な配置を調べる。この方法によって、タンパク質の形(立体構造)や内部構造を知ることができるが、水溶液中での動的な構造情報は得られない。なお日本蛋白質構造データバンク(PDBj)には、クライオ電子顕微鏡を用いた構造解析データ(水溶液中で取り得る安定な立体構造を示すデータ)も公開されている。
4.スパイクタンパク質、ヌクレオカプシドタンパク質
ウイルスゲノムにコードされる構造タンパク質。スパイクタンパク質は、宿主細胞表面に存在する受容体と結合し、細胞へ侵入するために働く。コロナウイルスの特徴であるウイルス粒子表面の王冠(コロナ)状の突起の本体。ヌクレオカプシドタンパク質は、ウイルスのゲノムRNAと結合し、ウイルス粒子の中心を構築する。
5.メインプロテアーゼ、RNA依存性ポリメラーゼ
ウイルスゲノムにコードされる酵素タンパク質。メインプロテアーゼは、ゲノムRNAから翻訳されたポリタンパク質を切断し、ウイルスタンパク質の成熟に関わる。RNA依存性ポリメラーゼは、ウイルスゲノムの複製に関わるRNA合成酵素。
6.分子動力学(MD)法
原子間に働く力を計算し、運動方程式を繰り返し解くことで、分子の動きを追跡する方法。分子動力学法の基礎の開発について、2013年のノーベル化学賞が授与されている。MDはMolecular Dynamicsの略。
7.インシリコ創薬
細胞生物学的、生化学的な手法を主とする治療薬候補化合物の探索に対して、コンピュータ(シリコンチップ)の中で行う探索をインシリコ(in silico)創薬と呼ぶ。
8.ABINIT-MP
FMO計算専用に開発されている、国産の量子化学計算プログラム。1999年の理論開発当初に中野達也博士(国立医薬品食品衛生研究所)が開発を開始し、現在は望月祐志教授(立教大学)を中心とする研究グループで開発を継続している。並列性能に優れ、研究室規模のサーバーから超並列のスーパーコンピュータまでを利用した高速計算が可能である。
共同研究グループ
理化学研究所 生命機能科学研究センター 制御分子設計研究チーム
チームリーダー 本間 光貴(ほんま てるき)
研究員 渡邉 千鶴(わたなべ ちづる)
技師 神坂 紀久子(かみさか きくこ)
客員研究員 原田 俊幸(はらだ としゆき)
研究員 高谷 大輔(たかや だいすけ)
研究員 大山 達也(おおやま たつや)
星薬科大学 薬学部
准教授 福澤 薫(ふくざわ かおり)
特任助手 川嶋 裕介(かわしま ゆうすけ)
大学院博士課程 半田 佑磨(はんだ ゆうま)
みずほ情報総研株式会社
チーフコンサルタント 加藤 幸一郎(かとう こういちろう)
研究支援
本研究は、日本医療研究開発機構(AMED)創薬等先端技術支援基盤プラットフォーム事業(BINDS)の支援を受けて行われました。
発表者
理化学研究所
生命機能科学研究センター 制御分子設計研究チーム
チームリーダー 本間 光貴(ほんま てるき)
星薬科大学 薬学部
准教授 福澤 薫(ふくざわ かおり)
報道担当
理化学研究所 広報室 報道担当
星薬科大学 総務部
AMED事業に関すること
日本医療研究開発機構
創薬事業部医薬品研究開発課

