2021-02-05 ,理化学研究所,京都大学,横浜市立大学,大阪大学
理化学研究所(理研)科技ハブ産連本部医科学イノベーションハブ推進プログラム(MIH)医薬プロセス最適化プラットフォーム推進グループの松本篤幸研究員、奥野恭史グループディレクター(MIH副プログラムディレクター、京都大学大学院医学研究科教授)、京都大学大学院医学研究科の荒木望嗣准教授、横浜市立大学大学院生命医科学研究科の寺山慧准教授、大阪大学蛋白質研究所蛋白質構造生物学研究部門の加藤貴之教授らの共同研究グループは、AI技術の一種である深層学習[1]と分子動力学(MD)計算[2]を組み合わせることで、クライオ電子顕微鏡(cryo-EM)[3]で計測される立体構造データのみから、タンパク質の運動性情報(柔らかさ)を直接抽出する新たな手法の開発に成功しました。
本研究成果は、タンパク質やDNAなどの生体高分子の運動性を解析する新たなアプローチを提供するものであり、生命科学の進展や医薬品開発への貢献が期待できます。
近年のcryo-EMの目覚ましい技術発展により、巨大かつ複雑なタンパク質の立体構造が明らかになってきています。しかし、それらの運動性を取得することは高度な計算や実験でも困難です。
今回、共同研究グループは、このようなタンパク質の運動性を、cryo-EMの計測データのみから原子レベルで簡便・迅速に推定できる画期的なAI「Dynamics Extraction From cryo-EM Map;DEFMap」を開発しました。これによって、これまで解析が困難であった生体高分子の運動性が明らかになり、機能メカニズムに対する新たな知見の発見につながると考えられます。また、本手法は超巨大なウイルス粒子などにも適用することが可能です。
本研究は、科学雑誌『Nature Machine Intelligence』オンライン版(2月4日付:日本時間2月5日)に掲載されました。

タンパク質の柔らかさ(運動性)を予測するAI:DEFMap
背景
生命現象を支える生体高分子は、3次元的な姿「立体構造」とその水中での揺らぎの様子「運動性」の両方の性質を持っています。立体構造は分子認識や酵素活性など個々のタンパク質が担う機能の基盤であり、運動性はタンパク質の機能を厳密に制御していることが知られています。こうした生体高分子の運動性を知ることは、機能メカニズムの正確な理解につながり、ひいては病態解明や医薬品開発を行う上で必要不可欠です。しかし、これまでタンパク質の運動性を解明するには、専門的な実験計測やスーパーコンピュータなどによる計算など高度な技術が必要であり、容易ではありませんでした。
例えば、近年発展が著しいクライオ電子顕微鏡(cryo-EM)は、単粒子解析[4]を通じて未知であった生体高分子の立体構造を原子~近原子分解能で次々と明らかにしています。一方、cryo-EMで解析対象となる生体高分子は一般に巨大かつ複雑であるため、それらの運動性情報(柔らかさ)を得ることは技術的に困難でした。
研究手法と成果
cryo-EMの単粒子解析での撮影試料は生体高分子溶液を瞬間的に冷凍して準備することから、得られる単粒子画像群には、溶液中での動的なゆらぎを反映したさまざまな構造のスナップショットが含まれます。そのため、これらの画像を収集・解析することで最終的に得られるcryo-EM データ(3次元密度マップ)には、暗に運動性に関わる情報が含まれている(隠れている)と考えられます。そこで共同研究グループは、このcryo-EMデータに含まれる「隠れた」運動性情報を上手く抜き出すためのAI「Dynamics Extraction From cryo-EM Map;DEFMap」を開発しました(図1)。
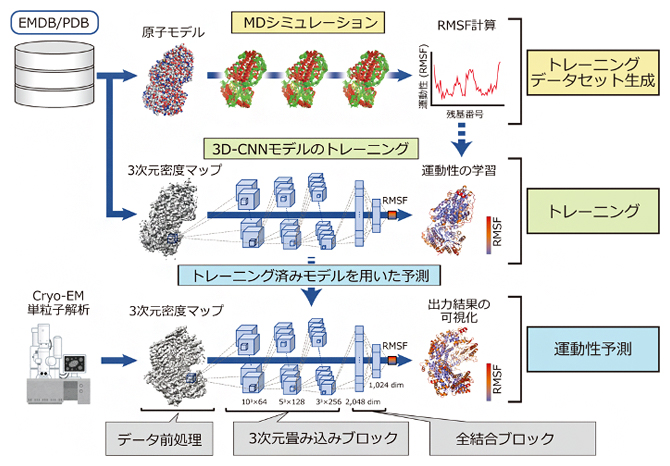
図1 DEFMapの全体的なワークフロー
公的データベースEMDB/PDBから選抜した生体高分子について、学習のための運動性情報を得るため原子モデルを用いたMD計算を行い、RMSF値を計算した(上段、トレーニングデータセット生成)。一方対応する3次元密度マップをサブボクセルに分解し、それぞれの位置の強度情報とMDで計算した運動性の情報との対応関係を学習した(中段、トレーニング)。この対応関係を学習したAIを用いることで、3次元密度マップの強度情報から、それぞれの位置における運動性を予測することができる(下段、運動性予測)。
具体的には、深層学習技術の一つである3次元畳み込みニューラルネットワーク(3D-CNN)[5]を用いて、3次元密度マップの局所的な強度パターン(強度情報)と、その位置に対応する原子の運動性情報を学習することでDEFMapを作成します。この学習を実現するには、タンパク質の正確な動的情報(タンパク質を構成する原子の運動性情報)が必要ですが、これまでのcryo-EM解析では直接、動的情報を得られません。そこで、公的データベースEMDB/PDB[6]から既にcryo-EM計測によって立体構造が解析された生体高分子25個を選出し、その立体構造モデルを用いた分子動力学(MD)計算を実施することで原子のゆらぎ(RMSF[7]値)を算出し、運動性情報としました(図1上段)。
次に、25個の生体高分子の各3次元密度マップを一辺15オングストローム(Å、1Åは100億分の1メートル)サイズのボックス単位(サブボクセル)に分解し、それぞれの単位ボックスの「3次元密度マップの強度情報」と「MD計算から得られる動的情報」を対応づけることで42万4930個の学習データセットを生成しました。そして、これらを学習することで生体高分子の運動性が予測できるAI「DEFMap」を作成しました。(図1中段)。ここで重要なことは、AIを作る(学習する)際にはMDで計算した動的情報が必要である一方、一旦学習してAIを作ってしまえば、あとは新たなタンパク質についてcryo-EMデータ(3次元密度マップ)のみを入力するだけで、その運動性情報を予測できることです。
続いて、DEFMapの性能を評価するために、学習に用いていない3種類のタンパク質の3次元密度マップ(テストデータセット)を入力して、DEFMapによって運動性を予測(図1下段)したところ、全てのケースにおいてMD計算で得られたRMSF値と良く一致する結果が得られ、またその結果は二次構造や分子内部などタンパク質の構造上の特徴を良く捉えていました(図2)。さらに、これらのタンパク質のMD計算には10~20時間の計算を要した一方、DEFMapを用いた予測は数分で完了したことから、本手法は運動性解析に関わる大幅なコスト削減を可能にします。
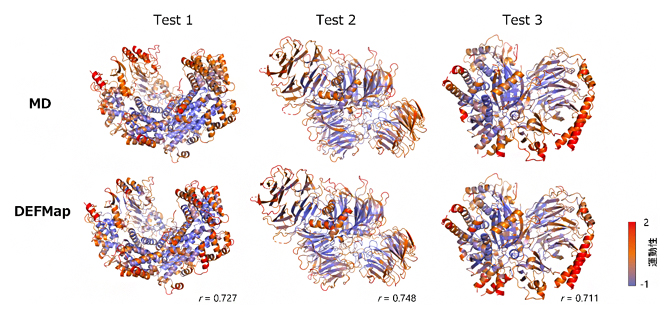
図2 MDで計算された運動性(上段)とDEFMapで推定した運動性(下段)との比較
三つのテストタンパク質の立体構造上にMDで計算された運動性(RMSF値の常用対数、上段)とDEFMapで推定された運動性(下段)を、右下に示すカラーバーに従って色分けした。赤が高い運動性、青が低い運動性を持つ領域であることを示す。それぞれのMDで計算した運動性とDEFMapで推定した運動性の類似度を相関係数rで右下に示す。両方の運動性が良く一致しているのが分かる。テストタンパク質のEMDB/PDB IDは、それぞれTest 1はEMD-4241/6FE8、Test 2はEMD-7113/6BLY、Test 3はEMD-20308/6PCVである。
また、これら3種のうちの一つのデータセット(図2のTest 3)については実験的に決定された運動性に関わるデータが公開されており、その結果とも良い一致が見られました(図3)。このことは、DEFMapが学習に用いていないタンパク質についても、3次元密度マップのみから局所の運動性を精度良く推定できることを示しています。
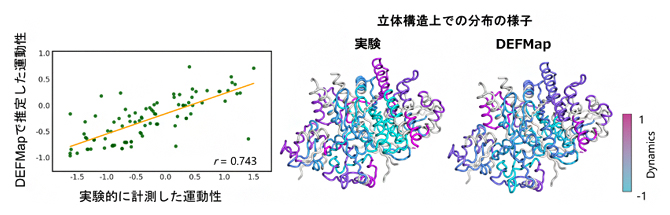
図3 実験で計測した運動性とDEFMapで推定した運動性との比較
図2のTest 3タンパク質について、公開されていたペプチドフラグメントレベルの動的情報を検証に用いた。左に、実験とDEFMapの運動性の相関図を示す。緑で各運動性の値を、オレンジでそれらの回帰直線を示す。また、右にそれぞれの運動性の立体構造上の分布をカラーバーに従って色分けした。紫が高い運動性、青が低い運動性を持つ領域であることを示す。実験とDEFMapの運動性が良く一致していることが分かる。
さらに、本手法の有用性を示すために、DEFMapを用いて生命現象に直接関与する運動性イベントの検出を試みました。生体内で起こる最も重要なイベントの一つとして「分子間相互作用」が挙げられます。例えば、医薬品はこの分子間相互作用を制御することで薬効を発揮します。一般に相互作用の際には、結合面が安定化されることが知られています。
そこで、相互作用分子(リガンド)の存在下/非存在下で決定された3次元密度マップをEMDB/PDBから新たに選抜し、それぞれの運動性を推定し比較しました。すると、リガンド存在下では、タンパク質においてリガンド認識に関わる領域の運動性に有意な低下が見られること(図4上段)、さらにその中でもリガンドとの重要な相互作用を担うアミノ酸残基の運動性が特に低下していることが示されました(図4下段左)。また興味深い観察結果として、相互作用部位とは離れた場所で構造状態の安定化を示す運動性が抑制されていること(アロステリック効果[8])が観察されました(図4下段右)。この領域の立体構造モデルを比較したところ、リガンドの有無にかかわらずほぼ同一であった(図4下段右スティックモデル)ことから、この知見はDEFMapにより3次元密度マップデータを直接解釈することで初めて得られたことになります。
DEFMapの入力データとして必要なものは3次元密度マップデータのみであることから、適用範囲が分子量や系の複雑さに制限されません。そのため、通常では運動性解析が困難な超巨大ウイルス粒子についても、迅速に分子全体の運動性を可視化することが可能です(図5)。図5から分かるようにウイルス表面の突起部分が赤くなっており、非常に運動性が高いことが分かります。新型コロナウイルス感染症でも問題視されているように、ウイルスの細胞への接着はこの突起部分が細胞の受容体に結合することで起こることから、この突起部分の運動性を定量的に見積もることで、細胞への接着のしやすさを評価できる可能性があります。
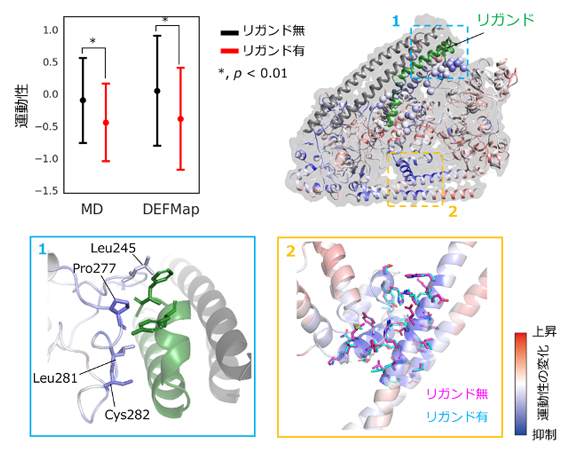
図4 リガンド結合に伴う運動性変化の検出
上段:左は、リガンド結合界面に位置する全アミノ酸残基の運動性の平均値(丸)と標準偏差(バー)を示すグラフ。MD計算、DEFMapともにリガンド結合に伴い、運動性の有意な低下が観察された。右は、タンパク質立体構造上で運動性の変化の分布を右下のカラーバーに従って示している。このうち緑色で示す部分がリガンドである。この可視化により、リガンド結合領域の残基で青色(運動性抑制)傾向が強いことが確認できる。
下段:全体図(上段右)の点線領域の拡大図。左は、リガンドとの重要な相互作用を担う四つの残基をスティックモデルで示している。右は、リガンド結合部位とは離れた場所で、運動性が抑制された残基側鎖をスティックモデルで示しており、立体構造モデルはリガンドの有無にかかわらず、ほぼ同一であった。
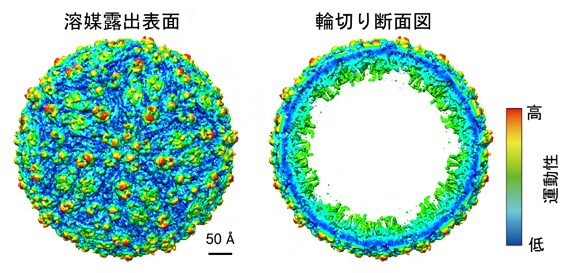
図5 超巨大ウイルス粒子(EMDB ID: EMD-8139)の運動性推定
推定した運動性の分布を右のカラーバーに従って色分けしている。左には溶媒に露出した表面を、右には輪切りにした断面を示している。またスケールバーは50Åの長さを示している。例えば、図2のタンパク質Test 1の長軸の長さが100Å程度なので、今回予測に用いたウイルス粒子が非常に大きいことが分かる。
今後の期待
本研究では深層学習(AI)・分子動力学計算(シミュレーション)・cryo-EMデータ(実験)を組み合わせることで生体高分子の運動性を得る手法を世界で初めて構築しました。今後、スーパーコンピュータ「富岳」などの利用を通じて大規模な動的情報を蓄積することで、本手法のさらなる精度の向上が見込まれます。
一般に運動性の計測には高度な実験設備や知識・経験が必要ですが、本研究で構築した学習済みモデルはリポジトリ上で公開されており、あらゆる研究者が3次元密度マップから簡便に運動性を抽出・確認できます注1)。
今日、cryo-EMによる構造決定例は増加の一途をたどっており、そこで決定される巨大かつ複雑な生体高分子の運動性解析が今後の命題になると考えられます。本研究はそれに対する全く新しいアプローチを提案するものであり、今後の生命科学、医学、薬学分野の研究開発の加速に大きく貢献すると期待できます。
注1)DEFMap: Dynamics Extraction From cryo-em Map(英語)
補足説明
1.深層学習
近年のAIの進展の中心的技術、英語名でdeep learningである。神経回路網を人工的に模したニューラルネットワークを多階層に結合することで、学習能力と表現力を高めた機械学習の一手法。従来の機械学習手法と比較してさまざまな分野、特に画像認識の分野で高い性能を示すことが報告されている。
2.分子動力学(MD)計算
ニュートンの運動方程式に従って相互作用する原子の位置や時間変化を計算することで、分子の動きをコンピューターシミュレーションで再現する方法。タンパク質の溶液中での動的振る舞いの解析などに利用されている。非常に精密な計算が可能であるが、計算コストがかかるためスーパーコンピュータを用いることがある。MDはMolecular Dynamicsの略。
3.クライオ電子顕微鏡(cryo-EM)
急速凍結した溶液試料を電子線照射により拡大撮影することで、試料の構造情報を得る手法であり、2017年にノーベル化学賞の受賞対象となった。電子直接検出器が実用化されたことで生体高分子の立体構造が原子~近原子分解能で観察できるようになり、急速な普及の結果、近年では本手法によって新しい立体構造が次々に報告されている。cryo-EMはcryo-electron microscopyの略。
4.単粒子解析
ここでは生体高分子を対象としたクライオ電子顕微鏡を用いた解析手法を指す。数万~数百万枚のさまざまな向きの単分子画像を収集し画像解析することで、3次元の密度マップを再構成することができる。
5.3次元畳み込みニューラルネットワーク(3D-CNN)
3次元オブジェクトに対するパターン認識や分類に広く利用されている深層学習技術の一つであり、MRI画像やCT画像などに応用されている。3D-CNNは3D- Convolution Neural Networkの略。
6.EMDB/PDB
PDBは、cryo-EMなどによって決定されたタンパク質などの生体高分子の3次元原子座標を蓄積している国際的な公共データベース。またEMDBは、cryo-EMによって得られた3次元密度マップを蓄積している国際的なデータベース。Cryo-EMの3次元原子座標は3次元密度マップに基づいて構築される。PDBはProtein Data Bankの略。EMDBはThe Electron Microscopy Data Bankの略。
7.RMSF
MD計算によって得られる各原子の揺らぎを、平均位置からのずれ(標準偏差)として数値化したもの。柔軟な領域は大きい値になり、反対に固い領域では小さい値になる。RMSFはroot-mean square fluctuationの略。
8.アロステリック効果
タンパク質の機能制御機構として見られる現象の一つで、ここではリガンド結合に伴う立体構造上の変化が、その結合部位から遠く離れた部位に伝わって構造特性の変化を誘起することを指す。
共同研究グループ
理化学研究所 科技ハブ産連本部 医科学イノベーションハブ推進プログラム(MIH)
医薬プロセス最適化プラットフォーム推進グループ
研究員 松本 篤幸(まつもと しげゆき)
グループディレクター 奥野 恭史(おくの やすし)
(MIH 副プログラムディレクター、京都大学大学院 医学研究科 教授)
京都大学大学院 医学研究科
准教授 荒木 望嗣(あらき みつぐ)
京都大学大学院 薬学研究科
大学院生 石田 祥一(いしだ しょういち)
横浜市立大学大学院 生命医科学研究科
准教授 寺山 慧(てらやま けい)
大阪大学 蛋白質研究所 蛋白質構造生物学研究部門
教授 加藤 貴之(かとう たかゆき)
研究支援
本研究は、文部科学省 ポスト「京」重点課題1「生体分子システムの機能制御による革新的創薬基盤の構築(代表者:奥野恭史)」、同 スーパーコンピュータ「富岳」成果創出加速プログラム「プレシジョンメディスンを加速する創薬ビッグデータ統合システムの推進(代表者:奥野恭史)」、日本学術振興会(JSPS)科学研究費補助金若手研究(B)「がん遺伝子産物Rasの動的な構造特性を介した機能発現メカニズムの解明(研究代表者:松本篤幸)による支援を受けて行われました。
原論文情報
Shigeyuki Matsumoto, Shoichi Ishida, Mitsugu Araki, Takayuki Kato, Kei Terayama, Yasushi Okuno, “Extraction of protein dynamics information from cryo-EM maps using deep learning”, Nature Machine Intelligence, 10.1038/s42256-020-00290-y
発表者
理化学研究所
科技ハブ産連本部 医科学イノベーションハブ推進プログラム(MIH) 医薬プロセス最適化プラットフォーム推進グループ
研究員 松本 篤幸(まつもと しげゆき)
グループディレクター 奥野 恭史(おくの やすし)
(MIH 副プログラムディレクター、本務:京都大学大学院 医学研究科 教授)
京都大学大学院 医学研究科
准教授 荒木 望嗣(あらき みつぐ)
横浜市立大学大学院 生命医科学研究科
准教授 寺山 慧(てらやま けい)
大阪大学 蛋白質研究所 蛋白質構造生物学研究部門
教授 加藤 貴之(かとう たかゆき)
報道担当
理化学研究所 広報室 報道担当

