
発達障害の発症メカニズムの解明と新たな治療法開発に期待
2021-06-09 九州大学,名古屋大学,慶應義塾大学,日本医療研究開発機構
概要
九州大学大学院医学研究院の中嶋秀行助教、中島欽一教授らの研究グループは、広島大学大学院統合生命科学研究科の今村拓也教授、名古屋大学大学院理学研究科・高等研究院の辻村啓太特任講師、慶應義塾大学医学部生理学教室の岡野栄之教授らとの共同研究により、神経発達障害レット症候群(※1)の原因因子であるmethyl-CpG binding protein 2(MeCP2)(※2)がマイクロRNA(miRNA)(※3)を介して神経幹細胞(※4)の分化を制御していることを発見し、そのメカニズムを明らかにしました。
レット症候群は自閉症、てんかん、失調性歩行、特有の常同運動(手もみ動作)を主徴とする進行性の神経発達障害です。MeCP2遺伝子の変異により発症することはわかっているもののその発症機序の詳細は不明でした。本研究グループは脳の発生過程において、MeCP2が神経幹細胞のニューロンへの分化を促進し、通常はニューロンの機能を支持するアストロサイトへの分化は抑制していることを明らかにしました。また、そのメカニズムについて調べた結果、MeCP2はmiR-199aというmiRNAを介して脳の発達に重要な骨形成因子(BMP)シグナル(※5)を抑制することで、神経幹細胞の分化を制御していることがわかりました。さらに、MeCP2遺伝子に変異をもつレット症候群患者由来のiPS細胞から作製した脳オルガノイド(※6)ではBMPシグナルの亢進とアストロサイトへの分化増加がみられ、これらがBMPシグナル阻害剤により改善できることが明らかになりました。以上の結果は、レット症候群患者脳では、神経幹細胞からニューロンやアストロサイトへの分化バランスが上手く制御されていない可能性を示しており、そのバランスの正常化によるレット症候群の新しい治療法開発へとつながることが期待されます。
本研究成果は、2021年5月18日(火)午前11時(米国東部標準時間)に国際学術雑誌『Cell Reports』に掲載されました。なお、本研究は文部科学省科研費、日本医療研究開発機構(AMED)、精神・神経疾患研究開発費、レット症候群支援機構の支援を受けました。
 (参考図)MeCP2は神経幹細胞の分化バランスを制御する野生型神経幹細胞では、MeCP2とDrosha複合体がPri-miR-199aをプロセシングし、正常なレベルのMature-miR199a-3pが産生される。Mature-miR199a-3pはBMP下流転写因子Smad1を標的とすることでニューロン分化とアストロサイト分化の適度なバランスを制御する(左図)。
(参考図)MeCP2は神経幹細胞の分化バランスを制御する野生型神経幹細胞では、MeCP2とDrosha複合体がPri-miR-199aをプロセシングし、正常なレベルのMature-miR199a-3pが産生される。Mature-miR199a-3pはBMP下流転写因子Smad1を標的とすることでニューロン分化とアストロサイト分化の適度なバランスを制御する(左図)。
一方、MeCP2欠損神経幹細胞では、MeCP2欠損のためDrosha複合体がPri-miR199aを正常にプロセシングできず、Mature-miR199a-3pが十分量産生されない。その結果、Smad1タンパク質の異常増加により、ニューロン分化が抑制され、アストロサイト分化優位へとバランスがシフトする(右図)。
背景
X染色体上に存在するmethyl-CpG binding protein 2(MeCP2)遺伝子の変異は、レット症候群をはじめ、自閉症、てんかん、統合失調症などを含めた様々な精神疾患・神経発達障害への関与が示唆されています。レット症候群は獲得された運動、言語能力の喪失、精神遅滞、自閉症傾向などを示す進行性の神経発達障害であり、神経系細胞特異的にMeCP2を欠損したマウスはレット症候群患者と類似の表現型を示すことから、MeCP2の神経系での機能が重要であることが示唆されているものの、レット症候群の表現型に関与する決定的な下流標的因子は未だに同定されておらず、レット症候群発症機序の全貌は不明なままでした。
我々の研究グループは以前、MeCP2がmicroRNA (miRNA) マイクロプロセッサーであるDrosha複合体(※7)と会合し、特定のmiRNA(miR-199a)のプロセシングを促進することを見出していました。また、レット症候群の死後脳ではmiR-199aの発現が減少しており、miR-199a欠損マウスはMeCP2欠損マウスと同様の表現型を示すことも明らかにしていました。しかし、以前の研究は、神経幹細胞から産生されたニューロンでのMeCP2の機能にのみ焦点を当てたもので、ニューロン以外にもアストロサイトなど、他の神経系細胞を生み出し、脳発生に重要な神経幹細胞における機能は不明のままでした。そこで、脳の発達過程におけるMeCP2及びmiR-199aの機能を明らかにすることで、MeCP2遺伝子の機能異常が原因で引き起こされる発達障害のさらなる発症メカニズムに迫ろうと考えました。
内容
研究グループは生後1日目の野生型及びMeCP2欠損、miR-199a欠損マウス脳から海馬を取り出し1細胞遺伝子発現解析を行いました。その結果、MeCP2欠損及びmiR-199a欠損海馬では共通してニューロン細胞集団が野生型海馬より減少している一方、アスロトサイト細胞集団は増加していることが明らかとなりました。この結果より、MeCP2欠損及びmiR-199a欠損マウス脳では神経幹細胞の分化バランスがニューロンからアストロサイトへシフトしている可能性が考えられました。そこで、胎生14日目の野生型及びMeCP2欠損、miR-199a欠損マウスから神経幹細胞を単離し、培養皿の上で分化誘導を行った結果、MeCP2欠損及びmiR-199a欠損神経幹細胞はニューロンへの分化が減少し、アストロサイトへの分化が増加していることが確かめられました。また詳細な解析により、miR-199aは脳の発達過程に重要な機能をもつBMPシグナルの下流転写因子Smad1をターゲットにすることで神経幹細胞の分化を制御していることを突き止めました。
次に、これまでマウスを用いて得られた結果と同様の現象が、実際のレット症候群患者の脳においても引き起こされている可能性を調べる目的で、レット症候群患者由来のiPS細胞から脳オルガノイドを作製し検討を加えました。その結果、レット症候群患者iPS細胞由来脳オルガノイドではアストロサイトへの分化が亢進しており、Smad1のタンパク質量も増加していることがわかりました。さらに、レット症候群患者iPS細胞由来脳オルガノイドへBMPシグナル阻害剤を添加することで、これらの表現型を改善できることを見出しました(図1)。
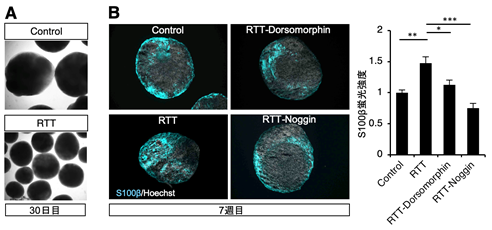
図1:培養30日目の正常(Contorl)及び変異MeCP2(RTT)を発現する脳オルガノイドの写真(A)。BMPシグナル阻害剤(Dorsomorphin,Noggin)を添加するとアストロサイトマーカー(S100β)の蛍光強度が減少する(B)。
このような結果から、MeCP2はmiR-199aを介して神経幹細胞の分化運命決定を制御しており、MeCP2/miR-199a/Smadという分子ネットワークがレット症候群病態において重要な役割を果たしていることが明らかになりました。
効果・今後の展開
今回の研究結果により、レット症候群の原因因子であるMeCP2が脳の発達過程において重要な役割を担っていることがわかると同時に、MeCP2変異を持つレット症候群の病態に新たな知見を提供しました。今回の成果を基に、今後は、BMPシグナル阻害による方法なども含めて、レット症候群を含めた発達障害の新規治療法開発につながることを期待しています。
用語解説
- (※1)レット症候群
- 自閉症やてんかん、失調性歩行、特有の常同運動(手もみ動作)を主徴とする進行性の神経発達障害である。X連鎖優性遺伝病であり、男性は胎生致死で女性のみが罹患する。レット症候群の80-90%にMeCP2遺伝子の変異がみられる。
- (※2)methyl-CpG binding protein 2(MeCP2)
- MeCP2の遺伝子はX染色体上に存在。MeCP2はメチル化された遺伝子のプロモーター領域に結合し、標的遺伝子の発現を抑制する転写抑制因子として同定されたが、私達の以前の研究によって、今回の研究でも重要な役割を果たすmiRNAの生合成にも関わることが明らかとなっている。MeCP2遺伝子の変異は、レット症候群の原因となるだけでなく、自閉症、双極性障害、認知障害、統合失調症患者にも認められる。
- (※3)マイクロRNA(miRNA)
- 細胞内に存在する長さ18から24塩基程度の1本鎖RNAであり、遺伝子の転写後発現調節に関与している。数百から数千塩基の一次前駆体(Primary-RNA)として転写され、核内でDrosha複合体にプロセシングされる。
- (※4)神経幹細胞
- 自己複製能および脳を構成する主要な細胞種であるニューロン、アストロサイト、オリゴデンドロサイトへの多分化能を併せもつ幹細胞。
- (※5)BMPシグナル
- BMPは、セリン/スレオニンキナ-ゼドメイン型受容体を介して、転写因子Smad1を含む下流因子を活性化することで細胞内にシグナルを伝達する。
- (※6)脳オルガノイド
- 多能性幹細胞(iPS細胞やES細胞)などを用いて、脳の形成過程を体外で模倣し、作製される三次元組織。
- (※7)Drosha複合体
- 一次前駆体として転写されたmiRNAを二次前駆体へとプロセシングするタンパク質の複合体。miRNAはその後核外へ移行し、別の酵素によってさらに、18から24塩基程度の成熟miRNAへとプロセシングをうける。
論文情報
- タイトル
- MeCP2 controls neural stem cell fate specification through miR-199a-mediated inhibition of BMP-Smad signaling
- 著者名
- Hideyuki Nakashima, Keita Tsujimura*, Koichiro Irie, Takuya Imamura, Cleber A. Trujillo, Masataka Ishizu, Masahiro Uesaka, Miao Pan, Hirofumi Noguchi, Kanako Okada, Kei Aoyagi, Tomoko Andoh-Noda, Hideyuki Okano, Alysson R. Muotri, and Kinichi Nakashima*(*共責任著者)
- 掲載誌
- Cell Reports, 2021
- DOI
- 10.1016/j.celrep.2021.109124
謝辞
本研究は文部科学省科研費(JP17H01390、JP16H06527、JP16K21734、JP16H06279(PAGS)、JP16J03827 and JP19K16918)、日本医療研究開発機構(AMED)革新的先端研究開発支援事業AMED-CREST「健康・医療の向上に向けた早期ライフステージにおける生命現象の解明」研究開発領域(JP20mg1310008)、精神・神経疾患研究開発費(27-7 and 30-9)、レット症候群支援機構(2019-01-09,10)の支援を受けました。
お問い合わせ
研究に関するお問合せ
九州大学大学院医学研究院 教授 中島 欽一 (なかしま きんいち)
名古屋大学大学院理学研究科・高等研究院 特任講師 辻村 啓太 (つじむら けいた)
慶應義塾大学医学部生理学教室 教授 岡野 栄之 (おかの ひでゆき)
報道に関するお問い合わせ
九州大学広報室
名古屋大学管理部総務課広報室
慶應義塾大学 信濃町キャンパス総務課:山崎・飯塚
事業に関するお問い合わせ
国立研究開発法人日本医療研究開発機構(AMED)
シーズ開発・研究基盤事業部 革新的先端研究開発課
革新的先端研究開発支援事業

