
2020-02-03 理化学研究所
理化学研究所(理研)生命機能科学研究センター細胞システム制御学研究ユニットのヴィピン・クマーリサーチアソシエイトと谷口雄一ユニットリーダーらの研究チームは、細胞の中にある複雑なゲノム[1]DNA(以下ゲノム)の構造様式の特徴を機械学習[2]により抽出する計算的手法を開発し、ゲノムを構成する新たな階層構造を発見しました。
本研究成果は、ゲノム上で起こる遺伝子発現の制御基盤の理解につながるものであり、iPS細胞[3]やオルガノイド[4]の効率的な作製や生命の発生機構の理解など、さまざまな生命・病理現象の基礎原理の理解に貢献すると期待できます。
生命の設計図であるゲノムは、細胞の核内で複雑に折り畳まれ、分化や発生時には、その構造状態や核内配置の変化を通じて遺伝子のスイッチオン・オフが切り替えられると考えられています。近年、数百万塩基対の長さでDNAがまとまった固まりを形成し、この構造が遺伝子の発現制御と密接な関わりがあることが分かってきました。一方、研究チームらの先行研究により、数百塩基対レベルでの単位構造も見いだされたことから、ゲノムのさまざまな階層での構造の特徴を抽出する手法の開発が求められていました。
今回、研究チームは、ゲノム領域を2分割する操作を繰り返しながら、ゲノム内に存在する幅広いサイズの構造を関連づけながら検出する計算アルゴリズムを開発しました。これを用いた解析の結果、遺伝子発現の活性と関連する新たな階層構造「エンクレーブ」を発見しました。
本研究は、科学雑誌『Nucleic Acids Research』オンライン版(2月3日付:日本時間2月3日)に掲載されます。

ゲノムに見いだされた階層構造「エンクレーブ」
背景
細胞内にあるゲノムDNA(以下ゲノム)にコードされた遺伝情報のおかげで、生物は生まれ、成長し、さまざまな環境に順応していくことができます。ゲノムは生物種ごとに固有な遺伝情報の総体です。ヒトの場合、約30億塩基対からなるDNAに約2万個の遺伝子がコードされています。細胞は、状況に応じてこれら遺伝子のオン・オフを切り替えることで、さまざまな機能を実現します。しかし、それぞれの遺伝子発現のオン・オフがどのように制御されているのか、その一般的な原理はまだよく分かっていません。
こうした遺伝子制御の原理を解き明かす一つの鍵になるものとして最近注目を集めているのが、ゲノムの構造様式です。特に、次世代シーケンサー[5]を用いた染色体立体配座捕捉法(Hi-C法)[6]が2009年に開発されたことで、細胞核の中でゲノムが実際にどのような構造をとっているかが網羅的に解析できるようになり、各遺伝子の制御状態と構造との密接な関連性が明らかになってきました。その結果、ゲノム構造は、数百万塩基対のDNAが空間的にまとまったTAD[7]と呼ばれるドメイン構造を軸に構成されており、遺伝子の発現はTADごとに制御されていることが分かってきました。
Hi-C法の技術開発は急速に進んでおり、今ではゲノム構造をさらに数百塩基対程度の細かさ(分解能)で解析できるレベルに達しています。これに伴い、これまでゲノムの基本構造として考えられてきたTADの内部にも、ドメインやループ[7]などの複雑な構造が存在していることが分かってきました。さらに研究チームは、2019年にHi-C法において世界最高分解能を達成し、ゲノム構造の最小単位であるヌクレオソーム[8](160~200塩基対)レベルでの構造決定を行なった結果、四つの隣り合ったヌクレオソーム同士が単位構造を形成していることを初めて見いだしました注1)。
これまでのHi-C法の技術開発では、連続したDNA領域で構成されるゲノム構造の検出に重点が置かれていました。一方、離れたDNA領域が空間的に接近している例は個別には発見されていますが、その全体像は不明なままとなっています。そこで本研究では、Hi-C法で得られたデータを用いて、ゲノムの全領域にわたる空間的なまとまりを検出することで、染色体レベルの大きな構造からTAD内部の小さな構造までを一括して見つけることができる計算アルゴリズムの開発を試みました。
注1)2019年1月18日プレスリリース「世界最高分解能で全ゲノムの3次元構造を解明」
研究手法と成果
本研究では、ゲノム上の遺伝子領域が近接してひと固まりになっている構造を「クラスター」と定義します。従来の方法では、ゲノム上のそれぞれの場所で、さまざまな長さの連続したDNA領域を想定し、領域内にあるDNAが全て空間的に近接していると認められた場合にクラスターと見なすやり方が用いられました。しかしこの方法では、連続していない領域間で見られる空間的なまとまりをクラスターとして検出できない、またサイズが大きく異なるクラスターを検出するのが難しいといった欠点が生じます。このため、TADのようなある程度決まった大きさのドメイン構造の検出は効率的に行えるものの、それ以外の構造の検出を行うのには向いていませんでした。
これに対して研究チームは、ゲノム全体の領域からスタートし、領域を繰り返し2分割していく方法を採用しました(図1)。こうすることで、大きなクラスター(染色体レベル)と小さなクラスター(数万塩基対レベル)の両方を、階層的に全てつなげながら検出できるようになります。さらにゲノム領域の分割にあたっては、機械学習の一つであるスペクトラルクラスタリング法[9]を利用しました。同法では原理的に、連続的ではない、飛び飛びのゲノム領域であっても一つのクラスターとして検出できるため、例えば異なる二つのゲノム領域が近接するループ構造も捉えることができます。研究チームはこの手法を、Hi-Cデータを2分割する(bisect)アルゴリズムの特徴から、「BHi-Cect法」と命名しました。
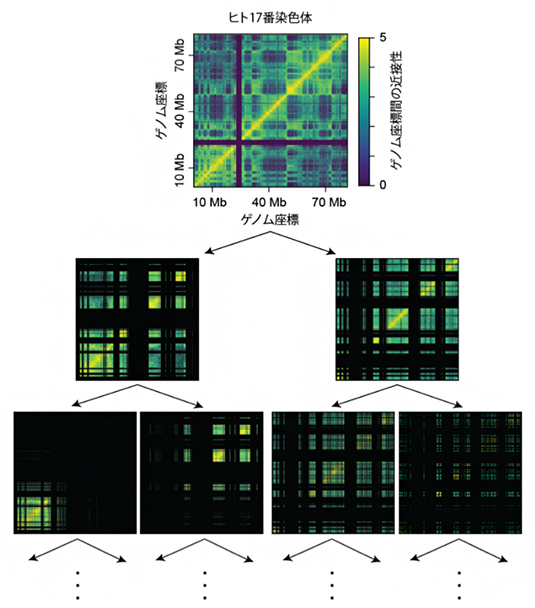
図1 BHi-Cect法によるヒト染色体構造のクラスター化
ヒト培養細胞に由来する17番染色体(約8,800万塩基対)を例として、クラスター構造を解析した。
(上)Hi-C法により得られたゲノム座標間の近接関係を表すグラフ。横軸と縦軸はいずれも、DNA配列を端から端まで順番に並べたときの位置(ゲノム座標)を示し、色はその領域のDNAが空間的に近い距離にある(黄色)か、離れているか(青)を表す。原点を通る対角線上(配列上で連続した領域)に100万塩基対(1Mb)程度の大きさで黄色の固まりが分断されている(くびれている)のが、TADと考えられる領域。なお、10Mbと40Mbの間にある黒い帯は、Hi-C法では近接関係が解析できなかった領域。
(中、下)上で示したHi-C法のデータを繰り返し分割することによって、ゲノム全域にわたって存在するさまざまなサイズのクラスター(緑色~黄色の長方形)が格子模様のように検出された。例えば中段のグラフでは、対角線上に位置する1Mb程度の長方形がTADに相当し、下段ではそれらがさらに小さいまとまりに分割されているのが分かる。また、分割にスペクトラルクラスタリングを用いることで、不連続な領域(対角線上から離れた領域)であっても一つのクラスターとして認識できる。
BHi-Cect法を用いてヒトゲノムのHi-Cデータの解析を行ったところ、それぞれのクラスターは、従来考えられてきたように連続したドメイン状の構造により構成されているのではなく、飛び飛びのゲノム領域の集合体により構成されていることが分かりました。そこで、検出したクラスターを、ドメインと区別する意味を込めて「エンクレーブ(enclave、入れ子構造の意)」と呼ぶことにしました。そして、BHi-Cect法で検出されたエンクレーブは、従来の研究で検出されてきたさまざまな構造(TAD、コンパートメント[7]、ループ)と、ゲノム上での境界の位置が一致していることも確認できました(図2)。
BHi-Cect法を用いてクラスターの分割を進めていくと、階層化が進んだ集積度の高いエンクレーブ構造を導出できます。図2で示した例では、1本の染色体がまず三つの大きなエンクレーブ(ひし形)に分割され、それらがさらに下層のエンクレーブ(正方形、円)で構成されていることが読み取れます。分割が進むほどエンクレーブ構造が占めるゲノム領域は小さくなっていきますが、面白いことに、分割中期に見出されるエンクレーブ構造(中程度エンクレーブ)に含まれる遺伝子の転写活性が全体的に高くなり、一方でそこから分割がさらに進行すると転写活性が平均以下まで低下することが分かりました(図3)。
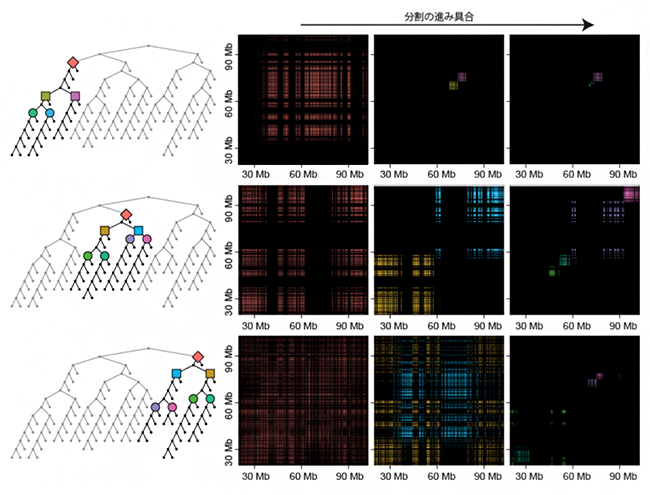
図2 分割の進展に伴うクラスター構造の変化
ヒト培養細胞に由来する15番染色体(約1億塩基対)について解析した結果。左の樹形図はエンクレーブの入れ子構造(ひし形>正方形>円)を表し、右のグラフは分割が進むごとに検出されるエンクレーブ(色は樹形図と対応)の領域を表す。分割の初期ではサイズの大きいエンクレーブが検出されるのに対し、中期(中)、末期(右)ではエンクレーブのサイズは小さくなる。
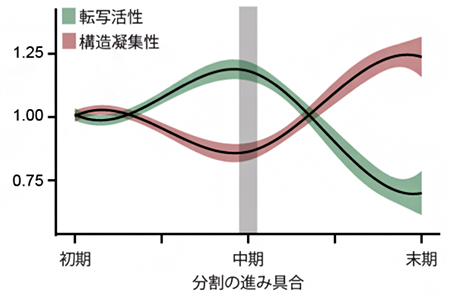
図3 分割の進展に伴う転写活性(緑)と構造凝集性(赤)の変化
分割の初期に現れる大きなエンクレーブ(大規模エンクレーブ)全体の凝集性と、そこに含まれる遺伝子の転写活性を平均したものをそれぞれ1とする。BHi-Cect法で得られた分割中期のエンクレーブ(グラフ中央、中程度エンクレーブ)では転写活性の上昇とゲノムの構造凝集性の低下が認められるのに対し、分割の末期(グラフ右側、末端エンクレーブ)では逆に転写活性の低下と凝集性の上昇が認められる。
この結果は、中程度エンクレーブが、異なる特徴を持つ2種類の領域で構成されていることを示しています。すなわち、中程度エンクレーブの表面にあるゲノム領域は比較的ほどけた構造でより高い転写活性を持つのに対し、内部には凝集性の高い末端エンクレーブが集中し、より低い転写活性を持つと考えると、うまく説明できます(図4)。エンクレーブの表面は露出しているためにさまざまな転写に関わるタンパク質の結合を受けやすくなり、逆に内部は覆われているためにタンパク質の結合を受けにくくなると推測されます。
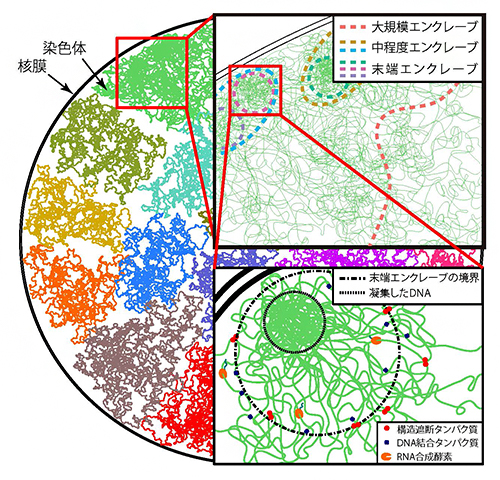
図4 本研究で提案したゲノム構造のモデル
ゲノム内にはさまざまな領域を集積したエンクレーブの階層構造が存在している。大規模、中程度、末端のエンクレーブをそれぞれ色分けした破線で表した。末端エンクレーブの境界より外側では、さまざまな因子が結合して転写が活性化されるのに対し、内側の特に凝集したDNA付近では、結合が抑制されて転写が不活性化される(右下図)。
今後の期待
今回、ゲノムの複雑な構造の中にあるさまざまな大きさの構造体を、階層的につなげながら検出するアルゴリズムを開発することで、遺伝子制御に関わる新たな構造単位「エンクレーブ」を見いだしました。今後、異なる細胞種や分化状態でのエンクレーブ構造を比較することにより、ゲノム構造と遺伝子発現の関係をより深く理解することが可能になると期待できます。
ゲノムが持つ配列だけでなく、その構造の意味が分かるようになると、ゲノムが行う複雑な遺伝子制御の仕組みを理解できるようになると考えられます。ゲノム構造から、発生機構の理解や生命・病理現象の基礎原理の理解を深めることで、iPS細胞やオルガノイドの効率的な作製法の開発、疾患診断技術や治療法の開発などの促進が期待できます。
補足説明
1.ゲノム
生命の遺伝情報を全て含んだデオキシリボヌクレオチド(塩基と糖が結合したもの)の重合体(DNA)。アデニン、チミン、グアニン、シトシンの4種類の塩基により構成されている。さまざまな遺伝子をコードした領域が並んでいる。
2.機械学習
膨大なデータをコンピュータに入力し、その中にある既知の特徴を繰り返しコンピュータに学習させるか、もしくはデータそのものからコンピュータに規則性を発見させることで、未知のデータに対する解答を自動で得る手法。
3.iPS細胞
脊椎動物の初期胚が持つ、全ての種類の体細胞へ分化する能力を多能性という。多能性を持ち、試験管内で培養して無限に増やすことができる細胞を多能性幹細胞という。iPS細胞(人工多能性幹細胞)は、皮膚や血液などから採取した細胞に少数の遺伝子などを導入して作製された多能性幹細胞である。
4.オルガノイド
幹細胞から人工的に作られる、生体内の組織または臓器に似た細胞組織体。
5.次世代シーケンサー
数百万から数億にわたる数のDNA断片の配列を並列して解読する技術。さまざまな生物種のゲノムを解読したり、RNA発現量を解析したりするのに用いられる。今日では生物学のみならず、医療・診断の分野にも幅広く普及しつつある。
6.染色体立体配座捕捉法(Hi-C法)
ゲノムの3次元構造の解析法の一つで、2009年に開発された。次世代シーケンサーを用いて空間的に近接したゲノム領域を網羅的に検出することで、ゲノムの3次元構造の解析を行える。Hi-Cはhigh-throughput chromosome conformation captureの略。
7.TAD、コンパートメント、ループ
細胞周期の間期の細胞核の中で、各々の染色体は100万塩基対ほどの球状に折り畳まれた構造を持ち、これをTAD(Topologically associating domain)と呼ぶ。TADはさらに折り畳まれ、遺伝子がよく転写されているTADが集まってAコンパートメントと呼ばれる領域を、転写されていないTADが集まってBコンパートメントと呼ばれる領域を核内空間に形成する。ループはDNAの折り畳み様式の一つで、離れた箇所にあるDNA領域を接近させることでループ状の構造を形成し、ループ内遺伝子とその外側の遺伝子の発現制御の境界となっていると考えられている。
8.ヌクレオソーム
細胞内におけるゲノムDNAの最小構造単位。ヒストンと呼ばれるタンパク質に、約150~200塩基対のDNAがおよそ一周半巻き付くことで形成される。
9.スペクトラルクラスタリング
互いに関連付けられたデータ要素をクラスターに分類するアルゴリズム。BHi-Cect法における解析の場合、データ要素はHi-C法が解析できるゲノムの単位領域に相当し、Hi-C法が導く領域間の近接関係の情報に基づいて二つのクラスターへの分類を行うことができる。
研究チーム
理化学研究所 生命機能科学研究センター 細胞システム制御学研究ユニット
リサーチアソシエイト ヴィピン・クマー(Vipin Kumar)
研修生(研究当時) シモン・ルクラーク(Simon Leclerc)
ユニットリーダー 谷口 雄一(たにぐち ゆういち)
(科学技術振興機構(JST) さきがけ兼任研究者(研究当時))
研究支援
本研究は、JST戦略的創造研究推進事業 さきがけ「統合1細胞解析のための革新的技術基盤(研究総括:浜地格)」、日本学術振興会(JSPS)科学研究費補助金挑戦的萌芽研究「真核細胞における遺伝子発現ノイズの制御機構」、同新学術領域研究「遺伝子発現の少数性生物学-少数分子による情報探索原理の解明-」、同若手研究A「高次生物における1細胞内全遺伝子発現の網羅的1分子計測」による支援を受けて行われました。
原論文情報
Vipin Kumar, Simon Leclerc and Yuichi Taniguchi, “BHi-Cect: A top-down algorithm for identifying the multi-scale hierarchical structure of chromosomes”, Nucleic Acids Research, 10.1093/nar/gkaa004
発表者
理化学研究所
生命機能科学研究センター 細胞システム制御学研究ユニット
ユニットリーダー 谷口 雄一(たにぐち ゆういち)
リサーチアソシエイト ヴィピン・クマー(Vipin Kumar)
お問い合わせ先
理化学研究所 生命機能科学研究センター センター長室 報道担当
山岸 敦(やまぎし あつし)
報道担当
理化学研究所 広報室 報道担当
及び無症状病原体保有者の発生について")
