標的とするメインプロテアーゼのウイルス複製機能阻害に求められるファーマコフォアをモデル化
2020-09-07 東京工業大学,筑波大学,日本医療研究開発機構
要点
- SARSウイルス(SARS-CoV)のゲノム情報とウイルス複製に寄与する酵素のメインプロテアーゼの立体構造の構造解析情報を元に、新型コロナウイルス(SARS-CoV-2)のメインプロテアーゼのウイルス複製機能阻害が期待される3化合物との複合体構造をモデル化
- 分子動力学シミュレーション(1)により、新型コロナウイルスのメインプロテアーゼの複製機能阻害に重要なファーマコフォア(2)のモデリングに成功
- 新型コロナウイルスのメインプロテアーゼ標的医薬品候補であるα-ケトアミド阻害剤で、本研究で求めたファーマコフォアの正しさを確認
概要
東京工業大学情報理工学院情報工学系の関嶋政和准教授を中心とする、同大学 物質・情報卓越教育院の安尾信明特任講師、筑波大学医学医療系生命医科学域の吉野龍ノ介助教の研究グループは、スーパーコンピュータTSUBAME3.0(3)を用いた分子動力学シミュレーションにより、新型コロナウイルス(SARS-CoV-2)の複製に関わる酵素「メインプロテアーゼ」の機能を阻害する治療薬の候補となる化合物が満たすべき、ファーマコフォアのモデリングに成功した。またメインプロテアーゼを標的とする医薬品候補であるα-ケトアミド阻害剤[参考文献]を用いて、このファーマコフォアが正しいことを確認した。
新型コロナウイルス感染症(COVID-19)の治療薬探索では、既存の上市薬もしくは治験の一部を通過した薬をベースに薬剤候補を探索するドラッグリポジショニングという手法が取られており、安全性や開発期間の短縮が期待されている。本研究で明らかにしたファーマコフォアは、既存薬の探索だけでなく、新規の薬候補化合物の探索にも応用できる点で重要な成果だといえる。
今後は、このファーマコフォアに基づき、シミュレーションと統計的機械学習などの人工知能(AI)や生化学実験を組み合わせたスマート創薬によって、新型コロナウイルス感染症(COVID-19)の治療薬候補となる具体的な化合物探索を目指していく。
本研究成果は2020年7月27日に国際科学誌『Scientific Reports』に掲載された。
* 本研究の一部は、国立研究開発法人日本医療研究開発機構(AMED)創薬等ライフサイエンス研究支援基盤事業 創薬等先端技術支援基盤プラットフォーム(BINDS)の課題番号JP20am0101112の支援を受けました。
研究成果
創薬には、十数年に渡る長い期間と3,000億円以上とも言われる膨大な研究開発費が必要であり、近年はこの費用が増加傾向にある。これまで、新規化合物獲得のための期間と費用を削減し、有望な薬候補化合物を効率的に探索するためにさまざまな手法、アプローチが開発されてきた。
新型コロナウイルス(SARS-CoV-2)は、2019年12月に中国・武漢で出現し、パンデミックを引き起こした。その後、新型コロナウイルス感染症(COVID-19)の感染者数は2020年8月初旬に1,800万人を超え、今も感染が拡大を続けるなか、世界中で治療薬の探索が続いている。従来、SARSウイルス(SARS-CoV)の複製に関わるメインプロテアーゼに対して、ペプチド様抗HIV-1薬が有効であることが報告されていた。SARS-CoVとSARS-CoV-2の間には密接な系統的関係があるため、これらのメインプロテアーゼは多くの構造的・機能的特徴を共有している(図1)。そのためペプチド様抗HIV-1薬はSARS-CoV-2でも、メインプロテアーゼを標的とする薬剤の候補として考えられているが、SARS-CoV-2のメインプロテアーゼの原子レベルでの作用機序はこれまで明らかになっていなかった。

図1 SARSウイルス(SARS-CoV)と新型コロナウイルス(SARS-CoV-2)のメインプロテアーゼのアミノ酸配列とタンパク質立体構造の比較。アミノ酸配列は96%同一。
本研究では、スーパーコンピュータTSUBAME3.0を用いて、1マイクロ秒の時間スケールでの分子動力学(MD)シミュレーションを実施することで、SARS-CoV-2のメインプロテアーゼと3種類の薬剤候補化合物との重要な相互作用を明らかにし、ファーマコフォアをモデリングすることに成功した。すべてのMDシミュレーションにおいて、SARS-CoV-2のメインプロテアーゼの41番目のHis(ヒスチジン)、153番目のGly(グリシン)、166番目のGlu(グルタミン酸)が、化合物の同じ官能基と相互作用を形成していた。この結果は、相互作用が、SARS-CoV-2のメインプロテアーゼに作用する薬剤の重要なターゲットであることを示唆している。
モデリングしたファーマコフォアの正しさを検証するため、SARS-CoV-2のメインプロテアーゼの機能をIC50=0.67±0.18μMで阻害することが知られているα-ケトアミド阻害剤が、このファーマコフォアに適合するか調べた。その結果、α-ケトアミドの1つの水酸基と2つのカルボニル基がファーマコフォアモデルと一致していることを確認した(図2)。
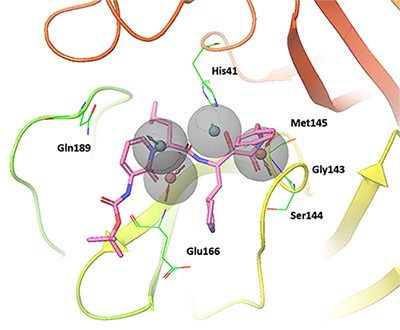
図2 SARS-CoV-2のメインプロテアーゼと医薬品候補であるα-ケトアミド阻害剤に関して、本研究でモデリングしたファーマコフォアへの適合を検証
SARSウイルス(SARS-CoV)には、メインプロテアーゼの145番目のCys(システイン)と共有結合する不可逆性阻害剤が数多く知られており、これらは競合阻害剤よりも高い結合親和性を有している可能性がある。しかし、COVID-19のような緊急性の高い疾患には、薬剤のリポジショニングが有効であり、本研究で提案したファーマコフォアは、Cysと共有結合を形成するために官能基を含まない化合物を評価することを可能にしている。
今後の展開
今後は、このファーマコフォアに基づき、シミュレーションと統計的機械学習などの人工知能(AI)や生化学実験を組み合わせたスマート創薬によって、COVID-19の治療薬候補となる具体的な化合物探索を目指していく。
参考文献
Zhang et al., Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors, Science 24 Apr 2020:Vol. 368, Issue 6489, pp. 409-412, DOI: 10.1126/science.abb3405
用語説明
- (1)分子動力学シミュレーション:
- 我々の体の中で、タンパク質やペプチドなどの生体分子は揺らぎながらその機能を果たしている。分子動力学シミュレーションは、タンパク質をつくっている原子や、周囲の水などの溶媒の動きを再現するシミュレーション手法である。分子動力学シミュレーションでは、原子ごとにニュートンの運動方程式を数値積分で解いていき、原子に働く力を求め、原子ごとの速度・座標の更新を行っていく。原子に働く力は、生体分子のシミュレーションの場合、1~2フェムト(10-15)秒ごとに求めることが多く、これを積み重ねてマイクロ(10-6)秒の時間スケールでのシミュレーションを行うため、TSUBAME3.0のような膨大な計算機資源が必要となる。
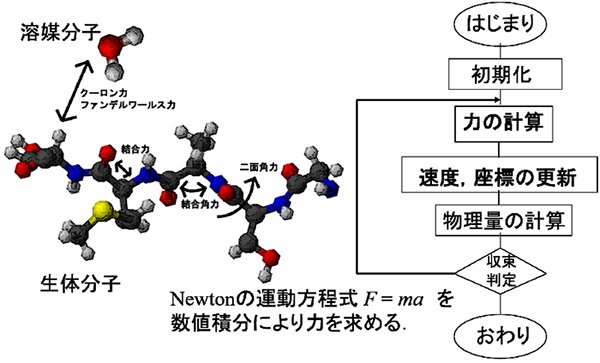
- (2)ファーマコフォア:
- 医薬品は、その標的となるタンパク質と相互作用することで、標的タンパク質の機能を阻害している。同じ標的タンパク質の同じ部位に結合する、医薬品による機能阻害に必要な官能基群の3次元配置をモデル化したものをファーマコフォアという。分子動力学シミュレーション等のシミュレーション手法に比べて、ファーマコフォアモデルを満足する化合物の探索は短時間で行うことが可能である。
- (3)スーパーコンピュータTSUBAME3.0:
- 東京工業大学学術国際情報センターが運用するスパコンで、2,160個の GPU を搭載し、12.15 ペタフロップスのピーク演算性能を持つ。最先端の研究教育の基盤として、広く学内外に計算資源を提供している。また産業利用にも大きく貢献している。
論文情報
- 掲載誌:Scientific Reports
- 論文タイトル:Identification of key interactions between SARS‑CoV‑2 main protease and inhibitor drug candidates
- 著者:Ryunosuke Yoshino, Nobuaki Yasuo, and Masakazu Sekijima
- DOI:10.1038/s41598-020-69337-9
お問い合わせ先
研究に関すること
東京工業大学 情報理工学院 情報工学系 准教授
関嶋 政和(せきじま まさかず)
筑波大学 医学医療系 生命医科学域 助教
吉野 龍ノ介(よしの りゅうのすけ)
取材申し込み先
東京工業大学 総務部 広報課
筑波大学広報室
AMED事業について
日本医療研究開発機構(AMED)創薬事業部医薬品研究開発課
創薬等ライフサイエンス研究支援基盤事業(BINDS)

