2022-11-02 理化学研究所,静岡県立総合病院,静岡県立大学
理化学研究所(理研)生命医科学研究センター ゲノム解析応用研究チームの寺尾 知可史 チームリーダー(静岡県立総合病院 免疫研究部長、静岡県立大学 特任教授)らの国際共同研究チームは、ヒトゲノムに数十万個存在するといわれるコピー数多型(CNV)[1]を従来の方法よりはるかに高感度に検出する手法を開発しました。
本研究成果は、現在の技術では遺伝子型決定が困難な遺伝子領域の遺伝的多型とこれに関連する形質への影響の解明につながると期待できます。
今回、国際共同研究チームは、世界で数千万人以上の規模でデータが存在するDNAマイクロアレイ[2](SNP[2]アレイ)のデータを用いて、祖先から継承された染色体の一部(セグメント)によってCNVを検出する「HI-CNV(Haplotype-Informed Copy-Number-Variation)」という手法を確立しました。HI-CNVでは、従来法(PennCNV[3])の6倍以上のCNVを検出しました。また、HI-CNVをUKバイオバンク[4]のデータに適用することで、CNVと56の量的形質[5]との関連を詳細に解析し、97の遺伝子座にわたる269の独立したCNV-形質関連を同定することに成功し、主要な関連を日本人の結果でも確認しました。今後、HI-CNVの枠組みを全エクソームシーケンス[6]データや全ゲノムシーケンス[7]データに拡張することで、DNAマイクロアレイデータでは網羅できなかったCNVの検出が可能になります。
本研究は、科学雑誌『Cell』オンライン版(10月27日付)に掲載されました。
背景
ヒトゲノムには、1細胞当たり通常2コピーの遺伝子が存在しますが、遺伝子のコピー数には個人差があり、ある個人によっては1コピーのみ(欠失)あるいは3コピー以上(重複)となり、これを「コピー数多型(CNV)」と呼びます。CNVは、精神神経疾患を含む多くのゲノム疾患の原因となることが知られています。
CNVはタンパク質をコードする遺伝子のコード配列に直接的に影響を及ぼし、タンパク質の機能喪失を引き起こすだけでなく、遺伝子量の増大や制御要素の欠損を引き起こし、間接的にコード配列の発現量、ひいてはタンパク質の発現量に大きく影響を及ぼします。従って、CNVが「形質」に与える影響を調べることは、形質への影響力を持つ新たな変異体を発見し、複雑な形質の遺伝的構造に関する理解を深める可能性を秘めています。
しかし、これまで、十分な検出力を持つフェノムワイドCNV関連解析[8]は、バイオバンク規模のコホート(集団)で利用できる低コストのDNAマイクロアレイから検出される大きなCNV(数十kb以上)の検討に限定されていました。
研究手法と成果
国際共同研究チームは、バイオバンクコホート内のハプロタイプ[9](祖先から継承された染色体)の共有を利用して、より感度の高いCNV検出法「HI-CNV(Haplotype-Informed Copy-Number-Variation)」を開発しました。
まず、Positional Burrows-Wheeler transform(PBWT)と呼ばれるアルゴリズムを用いて、2個体間で対立遺伝子が祖先と同じものを共有する状態のIBD(identity-by-descent)セグメントを迅速に特定し、各ゲノム位置において最も近い「haplotype neighbors」、すなわちコホート内の他のハプロタイプと最も長くマッチするIBDセグメントを特定しました(図1)。次に、個体の遺伝データからCNVが存在する可能性に関する定量的情報と、haplotype neighborsから対応する情報を利用して、共通祖先に由来するハプロタイプ上で共有されたCNVを隠れマルコフモデル[10]を用いて検出しました。
さらに、UKバイオバンクコホートで利用可能なSNPアレイの遺伝子型プローブ強度データにHI-CNVを適用するために、対立遺伝子特異的プローブ強度測定値をコピー数尤度(ゆうど)[11]に関する確率的情報に対応付ける確率的モデルを学習する方法を開発しました。CNV内の遺伝子型プローブは、CNV内にないプローブと比較して、特徴的な強度測定値を生成し、CNVを共有している複数の個体で一貫した偏差が観察されると、シグナルがより明確になることを利用しています。

図1 バイオバンクのSNPアレイデータからHI-CNVによりCNVを検出する基本的枠組み
従来のCNVを検出する標準的な手法では、個人ごとに別個にCNVに含まれるSNPの情報(黄色の部分)を用いてCNVを同定していた。これに対し、HI-CNVは、ある個体のSNPアレイデータと長い共有ハプロタイプ(水色の部分)を持つ個体(“haplotype neighbors”)の対応するデータとを一緒に解析して、CNVの検出率を向上させる。
HI-CNVをUKバイオバンクの登録者45万人に適用した結果、従来法(PennCNV)の6倍以上のCNVを検出しました(図2a)。43人の参加者の全ゲノムシーケンスパイロットデータを用いた検証分析では、HI-CNVの検証率は約91%とPennCNVと同等であり、正確性を保持したまま検出力が向上したことが確認されました(図2b)。さらに、バイオバンク・ジャパン[12]の登録者18万人にHI-CNVを適用したところ検出率は約93%と、UKバイオバンクと同様の性能であることが確認されました。これらHI-CNVの検出感度の上昇は、従来SNPアレイデータでは検出困難でありながら全CNVの大部分を占めていた、10kb以下のCNVに対する検出能力の向上によるものです(図2c)。
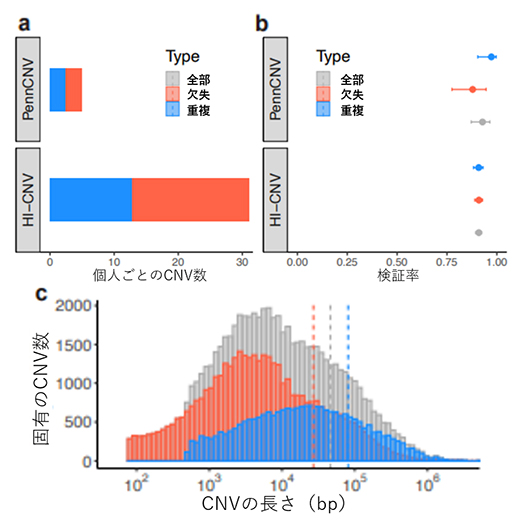
図2 従来法(PennCNV)から大幅に検出力を改善したHI-CNV
a)HI-CNVをUKバイオバンクデータに適用すると、従来法(PennCNV)の6倍以上のCNV(欠失・重複)を検出した。
b)全ゲノムシーケンスパイロットデータを用いた検証解析の結果。HI-CNVの検証率は約91%で、PennCNVと同等であった。
c)固有のCNV数とCNVの長さの関係。赤、灰色、青の破線は各CNVの長さの平均を示す。HI-CNVの検出感度は、10kb(104b)以下のCNVに対する検出力の向上によるものであった。
次に、検出された三つのカテゴリー(プローブレベル、遺伝子レベル、CNVレベル)のCNVと、身体測定形質、血圧、肺機能、骨密度、血球指標、血清バイオマーカーなど56の遺伝性定量形質の関連を調べました。線形混合モデルを用いて、UKバイオバンク登録者45万人について関連解析を実施し、さらにファインマッピング[13]を実施しました。その結果、97の遺伝子座における269のCNV-形質関連を特定しました(図3)。
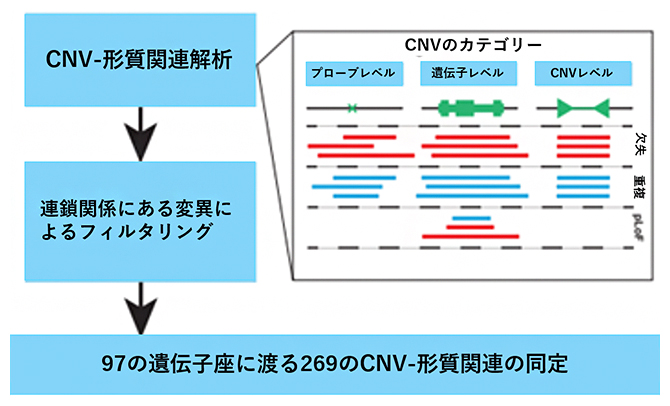
図3 CNV-形質関連解析のパイプライン
検出された三つのカテゴリー(プローブレベル、遺伝子レベル、CNVレベル)のCNVと、身体測定形質、血圧、肺機能、骨密度など合計56の形質との間で関連解析を実施し、さらにファインマッピングを実施することで97の遺伝子座における269のCNV-形質関連を特定した。
269のファインマッピングされたCNV-形質関連に関与する97の遺伝子座のうち、72の遺伝子座は新しいCNV-形質関連を示しました。72の新しい遺伝子座位の約半分である35座位については、標的遺伝子を同定できました。また、バイオバンク・ジャパンにおいて検出されたCNVを用いて、これら新しい遺伝子座位のうち形質などの条件が適切な14の関連に対して再現解析[14]を行ったところ、13の関連がその効果量についてUKバイオバンクと同じ方向性を示し、ほぼ一貫した効果量が観察されました。
今回同定された新しいCNV-形質関連遺伝子座の中には、(A)近傍(500kb以内)のどのSNPよりも強い関連を示す遺伝子座、(B)CNVが近傍のSNPとともに長い対立遺伝子系列を形成する遺伝子座、(C)推定上の標的遺伝子に新たに関与する追加遺伝子座が含まれていました。
例えば、E3ユビキチン-タンパク質リガーゼをコードするUHRF2遺伝子上の非常にまれなCNV(UKバイオバンク登録者19人が保有)は、身長の減少に関連していました(P=8.2×10-11)。UHRF2と身長との関連については、同じ遺伝子座のSNPはどれもゲノムワイドな有意性(P<5.0×10-8)を示していませんでした。また、骨形成タンパク質をコードするBMP5遺伝子の上流に位置する低頻度(MAF[15]=2.2%)の欠失は骨密度増加と強く関連しており(P=9.2×10-82)、近傍の強い関連を示すSNP(P=3.8×10-51)とともに長い対立遺伝子系列を形成することが示唆されました。これらの結果は、関連する欠失CNVをゲノムワイド関連解析(GWAS)[16]におけるファインマッピングに含めることの重要性を示しており、CNVとSNPを含む対立遺伝子系列のさらなる探索が必要であるといえます。
さらに、機能不明の遺伝子であるR3HDM4における非常に希少な欠失は、幼弱な網状赤血球数の増加と関連していました(P=3.5×10-11)。この関連性はR3HDM4遺伝子のPTV(proteintruncating SNP or indel variant)[17]でも確認され(P=2.7×10-7)、共通のイントロン(ゲノム上でタンパク質をコードしていない領域)SNPも網状赤血球数増加に対して強い有意性を示しました(P=6.6×10-86)。この欠失を詳しく調べると、エクソン(ゲノム上でタンパク質をコードしている領域)に重なるpLoF(predicted to cause loss of function)[18]欠失と、R3HDM4遺伝子の第1イントロン内に完全に位置するイントロン欠失の両方からなることが分かりましたが、いずれも網状赤血球数の増加に関連していることが判明しました。
これらの結果は、R3HDM4遺伝子のエンハンサー機能[19]が予測されるクロマチン領域[20]を含む欠失がまたがるイントロン領域が重要な制御的役割を担っていることを示唆しています。つまり、制御的CNVが形質に大きな影響を与え、時にはコーディング配列そのものに変化を与えるCNVと同じくらい強い影響を与える可能性があることを改めて示しています。
今後の期待
今回の研究では、新たな手法HI-CNVによりCNVの検出力が大幅に向上し、これに伴い72の遺伝子座にわたる多くの新しいCNV-形質関連を同定することができました。同定されたCNV-形質関連遺伝子座には、GWASによる既知のSNP-形質関連遺伝子座も含まれており、CNVが近傍のSNPよりもより強く関連している場合や、近傍のSNPとともに形質との強い関連を示す対立遺伝子系列を形成している場合があることが示されました。今後は、これらCNV-形質関連とSNP-形質関連を組み合わせて解釈することで、より正確な形質と関連遺伝子座の関係が解明されると期待できます。
また、今回HI-CNVの解析対象となったSNPアレイデータによって検出されたCNVは、通常ヒトゲノムに存在する数千のCNVのごく一部に過ぎません。今後、解析対象の枠組みを全エクソームシーケンスあるいは全ゲノムムシーケンスデータに拡張することで、現状では遺伝子多型の決定が困難な遺伝子座におけるCNVの検出が可能となり、さらなるCNV-形質関連が同定されると期待できます。
補足説明
1.コピー数多型(CNV)
ヒトのDNA配列を個々人で比較すると、塩基配列の違い(遺伝子多型)が見いだされる。遺伝子多型のうち、1kb以上の大きさの変異をコピー数多型という。同じ配列が繰り返される重複や、連続した配列が欠損する欠失などがある。タンパク質をコードする領域に生じれば、影響はより強くなる。CNVはcopy number variationの略。
2.DNAマイクロアレイ、SNP
DNAマイクロアレイは、基板の上に、遺伝的多型(主にSNP)に相補的なプローブを搭載したビーズを高密度に配置し、数十万~数百万の遺伝的多型を検出するための分析器具。SNPは一塩基多型と呼ばれ、変異が一つの塩基に限られるものを指す。SNPはsingle nucleotide polymorphismの略。
3.PennCNV
従来のDNAマイクロアレイを用いたCNV検出アルゴリズムの代表的プログラム。アレイのプローブシグナルを基に、個人ごとにCNVの検出を行う。
4.UKバイオバンク
英国で構築されているバイオバンクであり、50万人規模の疾患罹患情報、臨床情報、遺伝情報などから構成される。
5.量的形質
ヒトの身長や体重、胸囲などのように連続的、量的に変化する形質をいう。
6.全エクソームシーケンス
ゲノム中のタンパク質に関する情報を含むエクソン領域のみを配列解読すること。エクソン領域の一塩基多型(SNP)や、短い塩基の挿入(ゲノム配列の特定の位置に別の配列が挿入された形態)または欠失(ゲノム配列の一部が失われた形態)などを検出できる。疾患を引き起こす変異は、エクソン領域に多く存在していることが知られており、シーケンスの対象をエクソン領域に絞ることで、全ゲノムシーケンスと比べて低コストで重要な変異を検出できる。
7.全ゲノムシーケンス
全ゲノムDNAを鋳型として配列解読をすること。この配列解読によって、全ゲノム長の数倍~数十倍の総塩基数に相当するショートリードまたはロングリードデータが生成される。
8.フェノムワイドCNV関連解析
遺伝子多型(ここではCNV)を用いて、形質の変化と遺伝子多型の頻度差の関連を統計学的に検定する方法。多様な形質を対象として網羅的に解析するため、フェノムワイド(Phenome-wide)と呼ばれる。
9.ハプロタイプ
生物が持っている単一の染色体上の塩基配列のことをいう。父親由来、母親由来のハプロタイプを各個人が保有している。ジェノタイプ(遺伝子型)は多型の場所における塩基配列の組み合わせのため、ハプロタイプを決定するには統計学的なデータ解析が必要になる。
10.隠れマルコフモデル
観測された記号系列の背後に存在する状態の遷移系列を推測するために用いられるモデル。
11.尤度(ゆうど)
統計学における尤もらしさの程度。目の前の観察結果を基に、観測できないパラメータを推定する際に用いられる。
12.バイオバンク・ジャパン
オーダーメード医療実現化プロジェクトの基盤となるDNAサンプルや血清サンプルを47疾患(延べ約20万人)から収集し、臨床情報とともに保管している世界でも有数の資源バンク。
13.ファインマッピング
関連解析の手法の一つ。ある遺伝子座における、疾患に対する真の原因SNPの数を仮定した場合に、そのSNPが真に疾患の原因である確率を算出する手法。
14.再現解析
元の研究の基本的な発見が、研究の参加者や状況が異なる場合にも適用できるかどうかを判断するために、異なる状況や異なる被験者で、研究調査を繰り返すこと。
15.MAF
個々のヒトゲノムを比較すると、染色体上の場所が同一であっても、遺伝子や個々の塩基配列が異なる場合がある。これらの遺伝子や塩基配列をアレルという。ある染色体上の位置において、個人により異なる塩基配列を持つ場合、集団の中で2番目に高いアレルの頻度をMAFという。MAFはMinor Allele Frequencyの略。
16.ゲノムワイド関連解析(GWAS)
疾患の感受性遺伝子を見つける方法の一つ。ヒトのゲノム全体を網羅する遺伝子多型を用いて、疾患を持つ群と疾患を持たない群とで遺伝子多型の頻度に差があるかどうかを統計学的に検定する方法。検定の結果得られたP値(偶然にそのようなことが起こる確率)が低いほど相関が高いと判定できる。GWASは、Genome-Wide Association Studyの略。
17.PTV(proteintruncating SNP or indel variant)
遺伝子のコード配列を短縮することが予測される遺伝子変異(一塩基多型や挿入欠失による変異)のことをいう。
18.pLoF(predicted to cause loss of function)
遺伝子産物であるタンパク質の機能が低下または消失する、機能喪失型の変異(LoF)を引き起こすと推測される変異のことをいう。
19.エンハンサー機能
非翻訳領域の中で、遠位から遺伝子の転写の可能性を高める機能領域のことをいう。
20.クロマチン領域
核DNAとヒストンタンパク質の複合体。DNAがヒストンタンパク質に巻き付きコンパクトに折り畳まれている。遺伝子の転写がほとんど行われないヘテロクロマチン領域と活発に転写が行われるユークロマチン領域がある。
国際共同研究チーム
理化学研究所 生命医科学研究センター
ゲノム解析応用研究チーム
チームリーダー 寺尾 知可史(テラオ・チカシ)
(静岡県立総合病院 免疫研究部長、静岡県立大学 特任教授)
ハーバード大学 ブリガムアンドウィミンズ病院(米国)
助教授 ポールー・ロウ(Po-Ru Loh)
研究支援
本研究は、日本医療研究開発機構(AMED)難治性疾患実用化研究事業「シングルセル統合ゲノミクス解析が解き明かす強皮症の病態基盤の開発(研究代表者:寺尾知可史)」、同ゲノム医療実現推進プラットフォーム事業(先端ゲノム研究開発)「先天的/後天的構造多型に着目した免疫/精神疾患病態解明に関する研究開発(研究代表者:寺尾知可史)」、同革新的がん医療実用化研究事業「体細胞モザイクのがん発症および予後因子としての意義解明の開発(研究代表者:寺尾知可史)」の助成を受けて行われました。
原論文情報
Margaux L.A. Hujoel, Maxwell A. Sherman, Alison R. Barton, Ronen E. Mukamel, Vijay G. Sankaran, Chikashi Terao, Po-Ru LohMargaux L.A. Hujoel, Chikashi Terao, Po-Ru Loh, “Influences of rare copy number variation on human complex traits”, Cell, 10.1016/j.cell.2022.09.028
発表者
理化学研究所
生命医科学研究センター ゲノム解析応用研究チーム
チームリーダー 寺尾 知可史(テラオ・チカシ)
(静岡県立総合病院 免疫研究部長、静岡県立大学 特任教授)
報道担当
理化学研究所 広報室 報道担当
静岡県立総合病院 総務課
静岡県立大学 教育研究推進部 広報・企画室
")
