2023-09-28 京都大学iPS細胞研究所
ポイント
- ヒトiPS細胞由来腎オルガノイドを用いてアルポート症候群の病態を再現する疾患モデルを作製した。
- ヒトiPS細胞由来腎オルガノイドの疾患モデルはアルポート症候群患者の重症度による病態の差異を反映する。
- 特定の低分子化合物がアルポート症候群の原因である糸球体基底膜の変性を修復する可能性を見出した。
1. 要旨
平山 隆一郎 共同研究員(当時:CiRA増殖分化機構研究部門/大正製薬株式会社)、長船 健二 教授(CiRA同部門)らの研究グループは、ヒトiPS細胞由来腎オルガノイド注1)を用いてアルポート症候群の病態モデルを開発し、創薬候補物質の選択に有用であることを確認しました。
アルポート症候群は、糸球体基底膜注2)を構成するIV型コラーゲン注3)のα3(IV)、α4(IV)およびα5(IV)鎖をコードする遺伝子(COL4A3、COL4A4 およびCOL4A5 )の異常により引き起こされる遺伝性腎疾患であり、難聴や視力障害を伴います。これらの遺伝子異常により血液のろ過に重要な糸球体基底膜が脆弱化します。小児での発症後、比較的早期に末期腎不全に進行するため、患者さんの生活に大きな支障をきたします。現在、アルポート症候群に対する有効な治療法は少なく、新規治療法の開発が望まれています。一方、腎臓は複雑な組織構造からなり、培養細胞を用いて同疾患の病態を模倣することは困難でした。そのため創薬候補物質の探索に時間がかかっており、アルポート症候群の有効な新規治療法の開発には細胞培養による疾患モデルの新規作製が課題となっています。
本研究では、軽症型および重症型アルポート症候群の患者さん由来のiPS細胞と、軽症型のiPS細胞において原因遺伝子を正常型に修復したiPS細胞を作製しました。患者さん由来のiPS細胞をもとに、腎オルガノイドの作製方法を改良することで、アルポート症候群の病態を再現可能か検証しました。その結果、患者さん由来iPS細胞から作製した腎オルガノイドの糸球体基底膜において、α5(IV)の沈着が認められず、コラーゲンの構成が通常と異なることを確認しました。また、軽症型iPS細胞由来腎オルガノイドではα3(IV)が観察されるものの、重症型iPS細胞由来腎オルガノイドではα3(IV)が認められなかったため、重症度ごとの遺伝的特徴を反映していると考えられました。
さらに、タンパク質の構造異常を修正するとされる化合物である4-Phenylbutyric acid(4-PBA)を添加したところ、軽症型iPS細胞由来腎オルガノイドにおいてα5(IV)が免疫染色により検出可能となりました。以上より、本研究結果はアルポート症候群に対する治療コンセプトを提示するものであり、同技術はアルポート症候群の創薬候補物質の選択への応用も期待されます。
この研究成果は、2023年9月28日に英科学誌「Communications Biology」で公開される予定です。
2. 研究の背景
アルポート症候群は腎機能に重要な糸球体基底膜を構成するIV型コラーゲン遺伝子COL4A3、COL4A4 およびCOL4A5 の異常に起因し、20〜30代と比較的早期に末期腎不全に陥ります。その後は透析療法が必要となることから患者さんの生活を大きく制限し、費用も高額であることから経済的にも大きな負担となっています。降圧薬などが治療に有効であると報告されているものの、病態進行を完全に停止することは難しく、有効な治療法の開発が待ち望まれています。一方で、腎臓は血液をろ過するために複雑な構造からなり、その機能に重要な糸球体基底膜の異常などアルポート症候群の病態を細胞レベルで再現することは難しく、有効な新規治療法開発に向けた課題となっていました。
さまざまな組織に分化することのできるiPS細胞を用いて、種々の臓器を模倣したオルガノイドの作製方法が研究されています。これらのオルガノイドはいずれも臓器特徴的な機能や構造など従来の細胞株では不可能であった性質を再現できる可能性があります。
本研究では、アルポート症候群新規治療法の探索に有用な技術開発を目指し、アルポート症候群患者さんから疾患特異的iPS細胞を樹立しました。これらのiPS細胞から作製した腎オルガノイドはアルポート症候群様の病態を示しました。患者さん由来iPS細胞による腎オルガノイドをアルポート症候群の疾患モデルとして用いて、低分子化合物4-PBAが異常コラーゲンの構造を修正する可能性を見出し、新規治療法の探索に有益であることを確認しました。
3. 研究結果
1)腎オルガノイドの糸球体基底膜は培養時間依存的に成熟し、α5(IV)を発現する。
IV型コラーゲンは3本のα鎖で構成されており、各鎖が巻き付き合い三重らせん構造のトリマー(三量体)を形成します。生体において、α5(IV)は胎児期の未熟な糸球体基底膜には存在せず、臓器の発達に伴ってα3鎖、α4鎖およびα5鎖がヘテロトリマー(ヘテロ三量体)となり糸球体基底膜を構成することが知られています。α5(IV)を発現する腎オルガノイドの作製はアルポート症候群の病態を模倣するために必須です。
研究グループは、健常人由来iPS細胞を用いて腎オルガノイドの作製方法を検討することで、時間依存的にCOL4A5 遺伝子の発現が上昇し、その遺伝子産物であるα5(IV)が腎オルガノイドの糸球体基底膜様構造上に沈着することを確認しました(図1)。これらの結果は、腎オルガノイドが生体の腎臓の成熟した糸球体基底膜と類似していることを示しています。

図1 腎オルガノイドは時間依存的にCOL4A3、COL4A4 および
COL4A5 遺伝子発現が上昇し、α5(IV)が沈着する。
左:qRT-PCRによる経時的なCOL4A3、COL4A4 およびCOL4A 遺伝子発現解析。
右:腎オルガノイドの免疫染色。培養後、Day13+21においてα5(IV)の沈着がみられる。
2)アルポート症候群患者さん由来iPS細胞から作製された腎オルガノイドはα5(IV)を欠損する。
次に、COL4A5 遺伝子に変異のある軽症型および重症型のアルポート症候群の2名の患者さんからiPS細胞を樹立し、上記の方法を用いて腎オルガノイドを作製しました。
患者さん由来iPS細胞から作製した腎オルガノイドにおける糸球体基底膜様構造のα5(IV)を調べたところ、臨床病態と同様にタンパク質発現を欠損していました。一方で、軽症型のCOL4A5 遺伝子変異を正常型に修復したiPS細胞から作製した腎オルガノイドでは糸球体基底膜様構造にα5(IV)の発現を認めました。このことから、患者さん由来iPS細胞から作製した腎オルガノイドは、アルポート症候群の病態を再現していると考えられました。
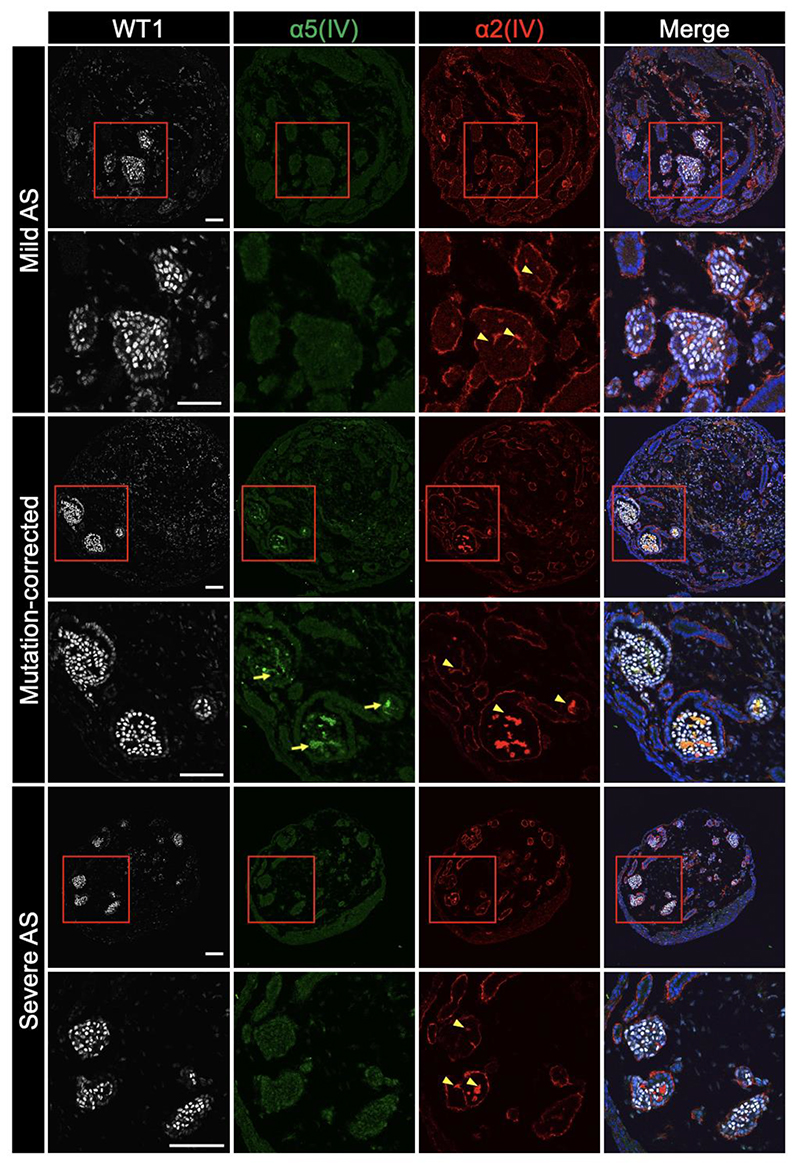 図2 アルポート症候群特異的iPS細胞から作製した腎オルガノイドは病態を模倣する。
図2 アルポート症候群特異的iPS細胞から作製した腎オルガノイドは病態を模倣する。
3)アルポート症候群患者さん由来iPS細胞から作製された腎オルガノイドは軽症型および重症型の特徴をそれぞれ反映する。
糸球体基底膜のⅣ型コラーゲンは、主にα3鎖、α4鎖、α5鎖が巻き付いたα3α4α5(IV)ヘテロトリマーにより構成されます。典型的な重症型のアルポート症候群患者さんではCOL4A5 遺伝子のみの変異であっても、α3α4α5(IV)ヘテロトリマーが形成されないためα5(IV)だけでなくα3(IV)およびα4(IV)も同様に糸球体基底膜から欠損します。一方で、非典型的な軽症型の患者さんでは、COL4A5 遺伝子変異を有するものの、α3鎖、α4鎖、そして構造異常を呈するα5(IV) がヘテロトリマーを形成し、糸球体基底膜に存在します。そのため、非典型的なアルポート症候群患者さんの糸球体基底膜では健康な人と同様にα3(IV)およびα4(IV)を検出可能です。
本研究で作製した腎オルガノイドも上記の特徴を反映しており、軽症型iPS細胞から作製した腎オルガノイドではα3(IV)が検出できる一方で、重症型では検出できませんでした(図3)。このことから、腎オルガノイドを用いてアルポート症候群患者さんの個人間の病態の差異を再現することができたと考えられます。
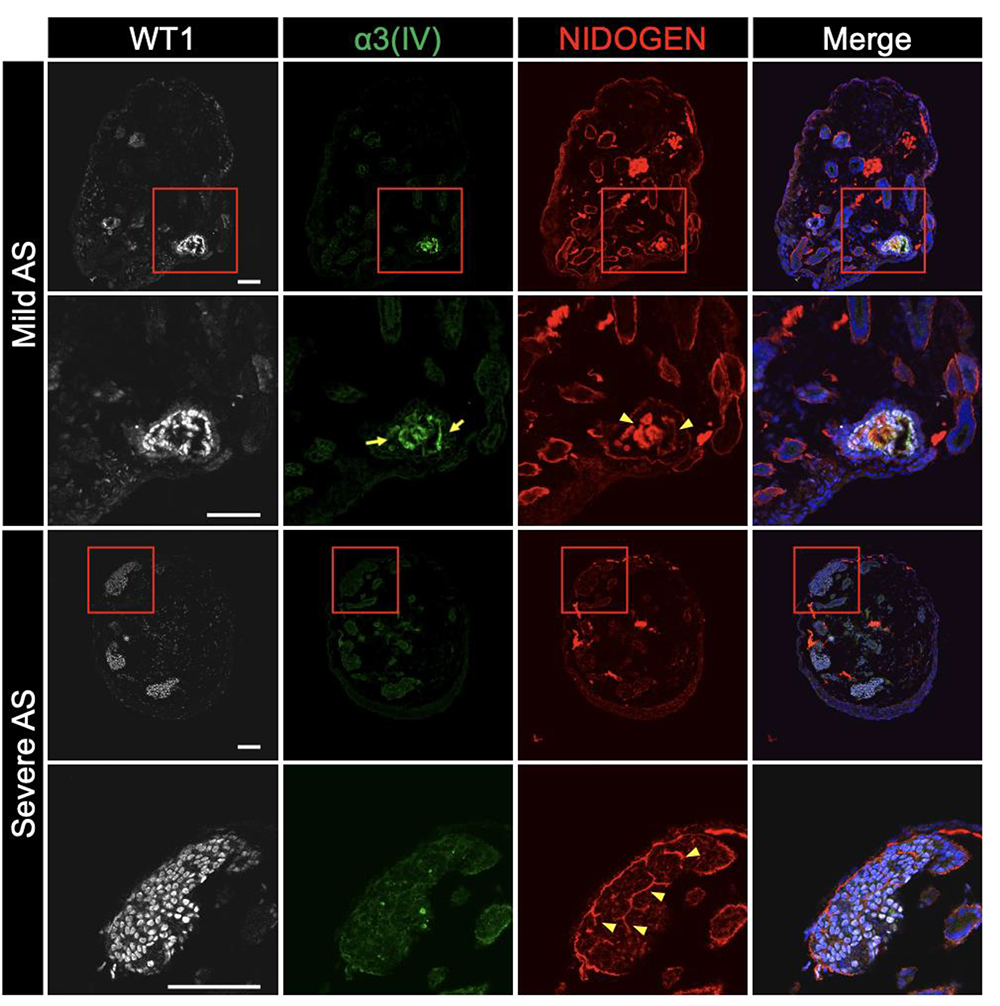
図3 アルポート症候群特異的iPS細胞から作製した腎オルガノイドは軽症型
および重症型アルポート症候群の病態をそれぞれ再現する。
4)低分子化合物4-PBAの添加によって軽症型iPS細胞由来腎オルガノイドのα5(IV)が検出可能となる。
タンパク質の構造異常は、特定の低分子化合物により修復できることが知られています。アルポート症候群のIV型コラーゲンの構造異常に対しても低分子化合物が作用する可能性を検証するため、軽症型iPS細胞から作製した腎オルガノイドに、構造異常を修復する可能性の報告されていた低分子化合物の4-PBA、TMAO、mannitolを添加し、α5(IV)の発現の有無を調べました。
その結果、4-PBA添加群においてα5(IV)を検出しました(図4)。この結果から、アルポート症候群においても、低分子化合物を用いてタンパク質の構造異常を修復する「ケミカルシャペロン療法」が新たな治療法となりうることが示唆されます。
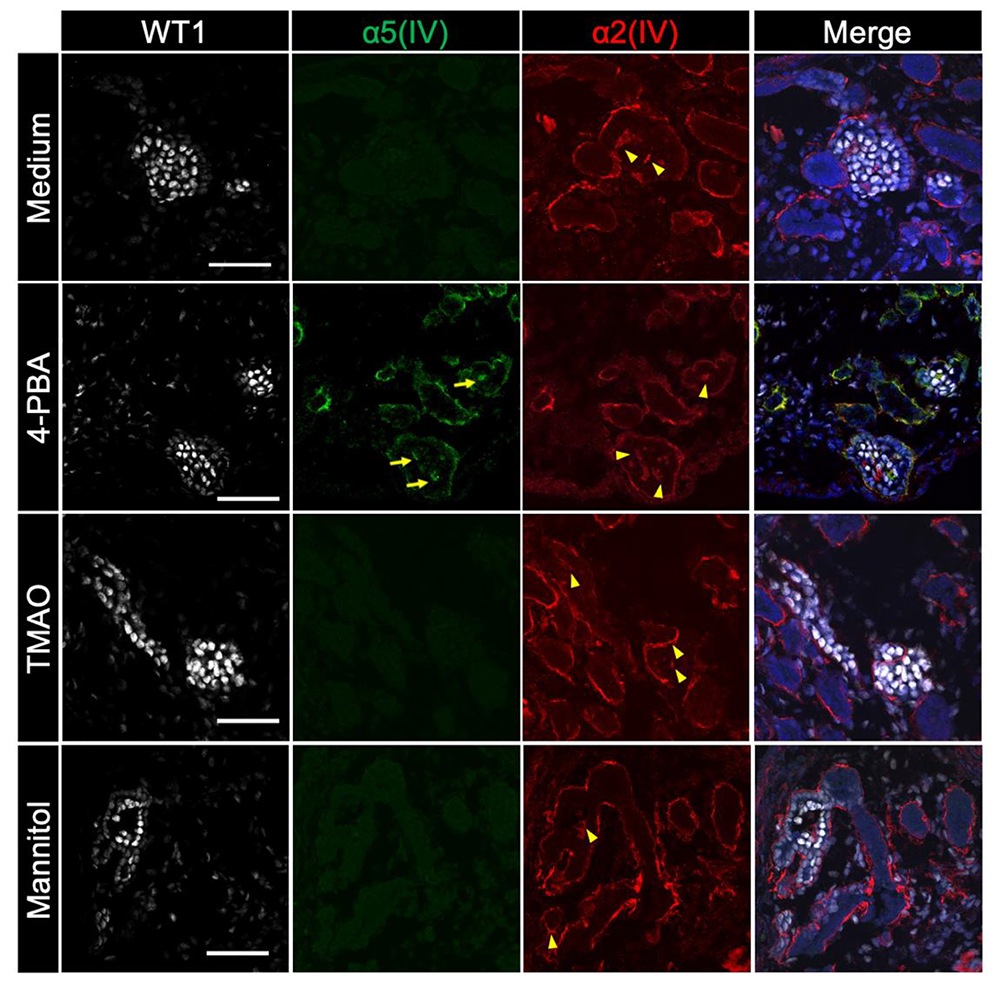
図4 低分子化合物4-PBAはα5(IV)構造異常を修復する可能性がある。
4. まとめと展望
本研究では、アルポート症候群の患者さんから樹立したヒトiPS細胞由来の腎オルガノイドが、アルポート症候群の病態を再現する培養系の疾患モデルとなることを示しました。さらに、今回開発した疾患モデルを使用して、タンパク質の構造異常を修復するケミカルシャペロン療法が有効である可能性を示しました。
本研究成果により、アルポート症候群を標的とした新規治療法の開発や病態の解明、非侵襲的な診断方法の開発が進み、患者さんが安心して受けることのできる新たな医療の実現につながることが期待されます。
5. 論文名と著者
- 論文名
iPSC-derived type IV collagen α5-expressing kidney organoids model Alport syndrome - ジャーナル名
Communications Biology - 著者
Ryuichiro Hirayama1,2, Kosuke Toyohara1, Kei Watanabe1, Takeya Otsuki1, Toshikazu Araoka1, Shin-Ichi Mae1, Tomoko Horinouchi3, Tomohiko Yamamura3, Keisuke Okita1, Akitsu Hotta1, Kazumoto Iijima3-5, Kandai Nozu3, and Kenji Osafune1,*
* : 責任著者 - 著者の所属機関
- 京都大学iPS細胞研究所(CiRA)
- 大正製薬株式会社
- 神戸大学大学院医学研究科 小児科学分野
- 兵庫県立こども病院
- 神戸大学大学院医学研究科 小児先端医療学分野
6. 本研究への支援
本研究は、下記機関より支援を受けて実施されました。
- 大正製薬株式会社
- 国立研究開発法人日本医療研究開発機構(AMED)[JP22bm0804013, JP23bm1123002]
- iPS細胞研究基金
7. 用語説明
注1)腎オルガノイド
iPS細胞などの多能性幹細胞から分化誘導を経て形成される腎臓様の構造体。尿細管様構造や糸球体様構造を有しており、胎児期の腎臓に類似するとされる。
注2)糸球体基底膜
腎糸球体において、糸球体毛細血管内皮細胞の基底膜と足細胞(糸球体上皮細胞)の基底膜が融合して形成され、コラーゲンやラミニンなどのタンパク質に富む。
注3)IV型コラーゲン
α1鎖からα6鎖までの6種類が知られており、COL4A1からCOL4A6 までの6遺伝子によりそれぞれコードされている。生体においては3本のα鎖がα1/α1/α2、α3/α4/α5またはα5/α5/α6の組み合わせでらせん構造を形成しヘテロトリマーとして存在する。

