
2020-08-07 理化学研究所,広島大学
理化学研究所(理研)生命機能科学研究センター発生動態研究チームの新海創也研究員、大浪修一チームリーダー、細胞システム制御学研究チームの谷口雄一チームリーダー、広島大学クロマチン動態数理研究拠点の冨樫祐一准教授(理研生命機能科学研究センター細胞場構造研究チーム上級研究員)らの共同研究グループは、ゲノム[1]構造データ(Hi-Cデータ)を高分子モデル[2]の4次元動態に変換する理論を構築し、Hi-Cデータ解析パイプライン[3]としてのシミュレーション手法「PHi-C法」を開発しました。
本研究成果は、細胞内におけるゲノムの動的状態や遺伝子発現制御機構の物理的理解につながり、ゲノム高次構造が持つダイナミクス制御機構とゲノム機能の関係の解明に貢献すると期待できます。
近年、ゲノムの3次元構造を調べる技術(Hi-C法[4])が急速に進展し、細胞状態に応じたゲノムの特徴的な折り畳みパターンと遺伝子発現のスイッチのオン・オフの関連が明らかになりつつあります。しかし、細胞の化学的固定[5]を必要とするHi-C法で得られるデータが、生きている細胞の中での動的なゲノム構造を反映しているかは不明でした。
今回、共同研究グループは、Hi-Cデータを解読してゲノムの4次元動態(3次元構造+1次元時間)に変換する理論を構築し、Hi-Cデータ解析パイプラインとしてのPHi-C法を開発しました。PHi-C法を用いることで、マウスES細胞(胚性幹細胞)[6]の多能性に重要なゲノム上の遺伝子領域の細胞核内における特徴的な動きや、染色体凝縮過程における棒状構造への経時的で動的な状態変化を、Hi-Cデータだけから再現することができました。
本研究は、科学雑誌『NAR Genomics and Bioinformatics』(6月号)に掲載されました。
背景
細胞内のゲノムDNAには、塩基配列の1次元パターンにさまざまな遺伝情報が書き込まれています。そのため、生物種ごとに固有な遺伝情報の総体としてのゲノムは生命の設計図と称されます。また、多細胞生物の体を構成する各細胞は、受精卵に由来する同一のゲノムを持っています。しかし、遺伝子の発現の仕方は細胞の状態や種類に応じて異なり、ゲノムの働き方は同一ではありません。ゲノムに書き込まれた遺伝情報がいつどのようにして適切に発現するのか、その仕組みはまだよく分かっていません。
そこで、近年注目されている技術が、ゲノム3次元構造を次世代シーケンサー[7]によって解析する「Hi-C法」です。この方法を用いた解析から、細胞核の中でゲノムは細胞状態に応じた特徴的な3次元構造をとり、遺伝子発現のスイッチのオン・オフを効率的に制御していることが分かってきました。しかしHi-C法で得られるのは、化学的に固定した100万個以上の細胞から抽出したゲノムの平均像であり、生きている細胞核の中での動的なゲノム状態を調べることができません。
さらに、最終的にHi-Cデータは2次元ヒートマップ[8]で表現されますが、その定量的情報が持つ物理的意義はよく分かっていませんでした。そのため、2次元Hi-Cデータを解読し、生きている細胞核内での4次元(3次元構造+1次元時間)ゲノム動態に関連付ける方法の開発が望まれていました。
研究手法と成果
Hi-C法では、ゲノムDNAとその結合タンパク質をホルムアルデヒド[5]で架橋固定[5]することで、空間的に近い距離にあるゲノム同士を連結させ、そのDNA断片ペアの塩基配列情報を、次世代シーケンサーを用いて網羅的に解析します。そして、100万個以上の細胞からの膨大なDNA断片ペアを解析することで、「ゲノム上のどの部分とどの部分が近接関係にあるかを意味する確率」という定量情報を得ることができます。このようなHi-Cデータは2次元ヒートマップとして表現され、その特徴的パターンは集団平均としてのゲノム3次元構造の特徴を反映します。
共同研究グループはまず、2次元Hi-Cデータの定量的意義を明らかにするため、ゲノム(1本の染色体)を「連結したビーズ」と見立てた単純な高分子モデルを使い、Hi-C法において検出されるゲノム間の空間的な近接効果を数式で記述しました(図1左)。その結果、従来考えられていたゲノム二点間距離(ゲノム上の二つの領域間の空間的な距離)とHi-Cデータにおける近接確率(ゲノム上の二つの領域が近接する確率)の間に厳密な対応関係は成立せず、その代わりに、「近接確率はゲノム二点間距離のばらつき度合いと関係する」という新しい数式を見いだしました。
さらに、この数式を高分解能のHi-Cデータに適用すると、ゲノム上の塩基対長さに対する近接確率の関係(近接確率曲線)に特徴的な振る舞いが出現し、その形状からHi-C法においてゲノム同士が連結する空間的距離が評価できることが予想されました。この予想を検証するため、共同研究グループの一人である谷口雄一チームリーダーらが開発したヌクレオソーム[9]レベルの高分解能のHi-C解析手法(Hi-CO法[10])注1)のデータを解析したところ、予想通り近接確率曲線に特徴的な形状が出現し、その形状からゲノム同士が連結する距離とヌクレオソームの大きさがほぼ同じであることが分かりました(図1右)。
この結果は、Hi-CO法がヌクレオソーム分解能でゲノム間連結を検出していること、および、今回理論的に見いだした数式が正しいことを支持するものです。
注1)2019年1月18日プレスリリース「世界最高分解能で全ゲノムの3次元構造を解明」

図1 Hi-C実験の高分子モデル化とHi-COデータ解析
左:連結したビーズとして表現できる高分子モデルの解析。ビーズでできたひもが折れ曲がったさまざまな構造の中から、二つのビーズの間の距離(二点間距離=dij)が近接する場合(距離σの範囲)だけを数理的に抽出できる(関数fσ(dij))。Hi-Cデータで得られるDNA断片ペアは、二点間距離のばらつき度合いを示す確率密度関数(p(dij))の中から、そのように抽出されたものとして数式で表現できる。これは、Hi-C実験において近接する二つの領域が化学的固定で連結されることに対応する。
右:理論解析の結果、近接確率曲線のゲノム距離が短いところに丸い形状が出現することが予想された。その予想通り、ヌクレオソーム分解能(160塩基)のHi-COデータには丸い形状が出現する。その形状は、ヌクレオソームの直径サイズbに対する近接距離σの比σ/bに依存する。データ解析の結果、その比の値は1に近い値であり、Hi-CO実験においてヌクレオソーム分解能でゲノム間近接が起きていたことが裏付けられた。
次に、細胞内のゲノムの振る舞いにより近い状況を再現するため、ネットワーク型相互作用を持つ高分子モデルを立てました。これは、連結されたビーズの全てのペアに、引力もしくは斥力の相互作用が働く動的なモデルです。このモデルに上記の数式を組み合わせた理論的な解析を行った結果、2次元Hi-Cデータと高分子モデルの相互作用パラメータ(ビーズペア間に働く力の変数)との間に成立する数学的な対応関係を発見しました。これは、ゲノム高次構造を反映した2次元Hi-Cデータにおけるあらゆるパターンが、ネットワーク型相互作用高分子モデルで再現できることを意味します。すなわち、2次元Hi-Cデータは、明確なゲノム3次元構造を直接意味するのではなく、ゲノム間の物理的な相互作用に対応し、その相互作用に基づいたゲノム動態と関係することが明らかになりました。
以上の結果から、Hi-Cデータを解読し高分子モデルの4次元動態に変換する理論を構築し、Hi-Cデータ解析パイプラインとしての「PHi-C(Polymer dynamics deciphered from Hi-C data)法」の開発に成功しました(図2)。PHi-C法では、2次元Hi-Cデータを入力すると、そのデータを90%以上の相関度合いで再現する最適な高分子モデルの相互作用パラメータが得られます。そして、その相互作用パラメータを用いることで、高分子モデルの4次元動態をシミュレーションすることや、ゲノム動態に関する理論曲線を計算することができます。
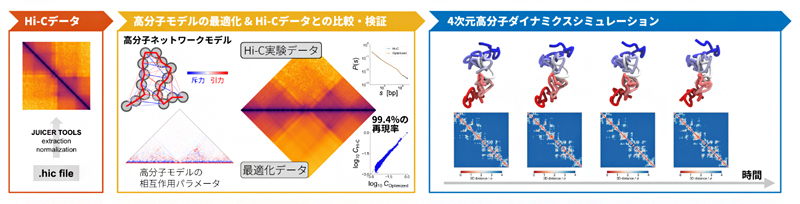
図2 Hi-Cデータを高分子モデルの4次元動態に変換するPHi-C法の流れ
PHi-C法では、2次元ヒートマップで表現されるHi-Cデータを入力すると、90%以上の相関度合いで入力Hi-Cデータを再現する高分子モデルの最適な相互作用パラメータを得ることができる。その相互作用パラメータを用いることで、入力Hi-Cデータに整合する高分子モデルの4次元動態をシミュレーションすることができる。
また、PHi-C法によって再現されるゲノム動態が、実際に顕微鏡で観察されるようなゲノムの動きをシミュレーションできるかどうか調べました。共同研究グループの一人である広島大学の落合博講師らはこれまでに、マウスES細胞の分化多能性の保持に重要なタンパク質(NanogとOct4)をコードしている二つのゲノム領域の動きには著しい違いがあることを、顕微鏡を用いた生細胞の経時観察により見いだしています注2)。PHi-C法を用いて、マウスES細胞のHi-Cデータを解析したところ、これら二つのゲノム領域の動きの違いを示すことができました(図3左)。さらに、その動きの差は、それらゲノム領域の局所的な構造的要因による違いであることも示しました(図3右)。
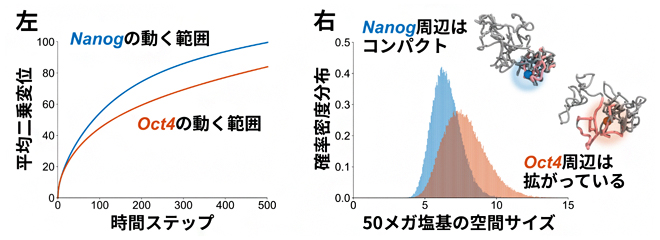
図3 マウスES細胞のHi-Cデータ解析
左:マウスES細胞の分化多能性に重要な二つのタンパク質(NanogとOct4)をコードするゲノム領域の細胞核内における動きは、Nanog遺伝子領域の方がOct4遺伝子領域に比べて動きが大きいことが報告されている。PHi-C法の解析の結果、Hi-Cデータだけから同様の振る舞いを計算することができた。平均二乗変位は、運動する物体の始めから終わりまでの動く範囲を表す指標で、値が大きいほど動きが大きい。
右:二つのゲノム領域周辺50メガ塩基対(5000万塩基対)が形成するゲノム構造の空間サイズを計算すると、相対的に、動きの大きいNanog遺伝子周辺はコンパクトな、動きの小さいOct4遺伝子周辺は広がった局所構造を形成していることが分かった。
次に、染色体レベルのゲノム動態への適用を検証するため、有糸分裂[11]時におけるニワトリBリンパ細胞のHi-Cデータを解析しました。これについても、顕微鏡で観察される一般的な染色体の形状変化の通り、染色体凝縮過程における棒状構造への経時的で動的な状態変化を再現できました(図4)。
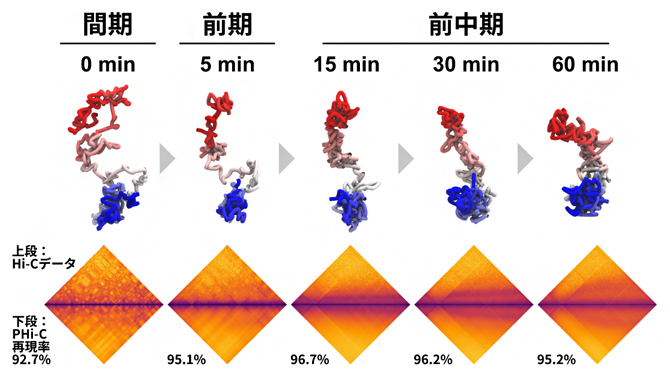
図4 間期から前中期におけるニワトリBリンパ細胞のHi-Cデータ解析
PHi-C法は、分裂前の間期から前中期に向かう有糸分裂時のニワトリBリンパ細胞のHi-Cデータを90%以上の相関度合いで再現した(下段のヒートマップの比較)。そして、4次元動態シミュレーションを行うと、間期における広がった形から顕微鏡で観察されるような棒状構造へ、経時的に状態変化する染色体凝縮過程を再現できた。
注2)Ochiai H., Sugawara T. and Yamamoto T. (2015) Simultaneous live imaging of the transcription and nuclear position of specific genes. Nucleic Acids Res., 43, e127
今後の期待
本研究成果によって、固定された細胞内での膨大なゲノム構造の特徴を反映したHi-Cデータと、生きている細胞内でのゲノム動態を関連付けることが可能になりました。この理論を応用したPHi-C法は、特別なコンピュータを必要とせず、Hi-Cデータと整合するようなゲノムの動く姿をシミュレーションするHi-Cデータ解析パイプラインです。
今後PHi-C法が普及することで、細胞内におけるゲノムの動的状態や遺伝子発現制御機構の物理的理解につながると考えられます。そして、ゲノム高次構造が持つダイナミクス制御機構とゲノム機能の関係の解明に貢献すると期待できます。
PHi-C法を応用したゲノムレオロジー解析技術の開発とPHi-C法の今後の展開については米国生物物理学会による学術雑誌『Biophysical Journal』(5月5日号)に掲載されました注3)。
なお、PHi-C法の解析コードは、soyashinkai/PHi-Cのホームページ![]() から利用できます。
から利用できます。
注3)Shinkai S., Sugawara T., Miura H., Hiratani I. and Onami S. Microrheology for Hi-C Data Reveals the Spectrum of the Dynamic 3D Genome Organization. Biophys. J. 2020;118(9):2220-2228.
補足説明
1.ゲノム
生物の染色体に含まれる全遺伝情報。アデニン(A)、チミン(T)、グアニン(G)、シトシン(C)の4種類の塩基によって構成されるDNA塩基配列に、さまざまな遺伝子をコードした領域が並んでいる。塩基配列のパターンは1次元文字列として表現できる。実体としてのゲノムDNAは、細胞内において3次元構造を持つ。そして、細胞内でその3次元構造が動くという観点として時間軸を加えることで、生きている細胞内でのゲノムは「4次元ゲノム動態」としての実体がある。
2.高分子モデル
染色体のような生体高分子の構造や動きを物理的に記述し、計算機上でシミュレーションするためには、そのモデル化が必須である。一般的に高分子はユニット分子が連結したものである。それゆえ、ユニット分子をある大きさを持ったビーズと見なし、その連結の仕方を物理的な相互作用として記述することによって、対象高分子をモデル化することができる。
3.パイプライン
次世代シーケンサーなどから得られるゲノム配列情報など、大量のデータを効率良く解析するための計算手法。
4.Hi-C法
3C(Chromosome Conformation Capture; 染色体立体配座捕捉)法を発展させた全ゲノム解析手法。細胞核内ゲノム3次元構造において空間的に近接する任意のゲノムDNA断片のペアを、次世代シーケンサーを用いて網羅的に検出し、ゲノム3次元構造を推定・解析できる。Hi-CはHigh-throughput chromosome conformation captureの略。
5.化学的固定、ホルムアルデヒド、架橋固定
化学的固定とは、生体試料の腐敗や分解を防ぐために化学的な処理を行うこと。ホルムアルデヒド水溶液(ホルマリン)は一般的な固定液で、高分子内のアミノ基同士を結合させる(架橋)ことで固定する。
6.ES細胞(胚性幹細胞)
哺乳類生物の発生初期の胚盤胞期に、胚盤胞内細胞塊から樹立された、多分化能を持つ培養細胞株のこと。多能性を持つ幹細胞には、ほかにiPS細胞(人工多能性幹細胞)がある。
7.次世代シーケンサー
数百万から数億にわたる数のDNA断片の配列を並列して解読する技術。さまざまな生物種のゲノムを解読したり、RNA発現量を解析したりするのに用いられる。今日では生物学のみならず、医療・診断の分野にも幅広く普及しつつある。
8.2次元ヒートマップ
行列のような2次元配列の各要素に値が格納されているデータに対して、各要素の値を色のグラデーションに対応させて可視化したグラフのこと。Hi-Cデータは二つのゲノム座標間の近接確率行列データであるため、その可視化には2次元ヒートマップが使われる。身近な例では、雨雲レーダーによって地図上の降水量が可視化される。
9.ヌクレオソーム
真核生物の細胞核内におけるゲノムDNAの最小構造単位。ヒストンと呼ばれるタンパク質の八量体に、約150~200塩基対のDNAがおよそ一周半巻き付くことで形成される。
10.Hi-CO法
単一ヌクレオソーム分解能で、さらにそれぞれの配向を含めたゲノム3次元構造解析を行う手法。真核生物のDNAは、ヌクレオソームが数珠状に連なったヌクレオソーム繊維を形作る。Hi-CO法は2019年当時世界最高分解能のHi-C法を実現した。Hi-COはHi-C with nucleosome Orientationの略。
11.有糸分裂
真核生物の細胞核の一般的な分裂の仕方。分裂の過程は前期・前中期・中期・後期・終期に分けられる。
共同研究グループ
理化学研究所 生命機能科学研究センター
発生動態研究チーム
研究員 新海 創也(しんかい そうや)
(研究開始時:広島大学 クロマチン動態数理研究拠点 特任助教)
チームリーダー 大浪 修一(おおなみ しゅういち)
細胞システム制御学研究チーム
チームリーダー 谷口 雄一(たにぐち ゆういち)
広島大学 クロマチン動態数理研究拠点
准教授 冨樫 祐一(とがし ゆういち)
(広島大学大学院 統合生命科学研究科 准教授)
(理化学研究所 生命機能科学研究センター 細胞場構造研究チーム 上級研究員)
特任助教 中川 正基(なかがわ まさき)
(現:大阪大学大学院 情報科学研究科 特任助教)
特任助教 菅原 武志(すがわら たけし)
(現:東京大学大学院 医学系研究科 特任助教)
広島大学大学院 統合生命科学研究科
講師 落合 博(おちあい ひろし)
東京大学 定量生命科学研究所
講師 中戸 隆一郎(なかと りゅういちろう)
研究支援
本研究は、日本学術振興会(JSPS)科学研究費補助金新学術領域(研究領域提案型)「分子修飾情報を実装した染色体数理モデルによるクロマチンドメイン内相互作用の研究(研究代表者:新海創也)」「染色体3次元構造理論の構築と応用:深層学習を援用した染色体4Dシミュレーション(研究代表者:新海創也)」「シンギュラリティ細胞の同定と解析のためのインフォマティクス技術の開発(研究代表者:大浪修一)」、科学技術振興機構(JST)戦略的創造研究推進事業CREST「科学的発見・社会的課題解決に向けた各分野のビッグデータ利活用推進のための次世代アプリケーション技術の創出・高度化(研究総括:田中譲)」における「データ駆動型解析による多細胞生物の発生メカニズムの解明(研究代表者:大浪修一)」による支援を受けて行われました。
原論文情報
Soya Shinkai, Masaki Nakagawa, Takeshi Sugawara, Yuichi Togashi, Hiroshi Ochiai, Ryuichiro Nakato, Shuichi Onami, “PHi-C: deciphering Hi-C data into polymer dynamics”, NAR Genomics and Bioinformatics, 10.1093/nargab/lqaa020
発表者
理化学研究所
生命機能科学研究センター 発生動態研究チーム
研究員 新海 創也(しんかい そうや)
チームリーダー 大浪 修一(おおなみ しゅういち)
細胞システム制御学研究チーム
チームリーダー 谷口 雄一(たにぐち ゆういち)
広島大学 クロマチン動態数理研究拠点
准教授 冨樫 祐一(とがし ゆういち)
報道担当
理化学研究所 広報室 報道担当
広島大学 財務・総務室 広報部広報グループ

