
2020-11-10 理化学研究所
理化学研究所(理研)生命機能科学研究センター計算分子設計研究チームの小松輝久研究員、沖本憲明上級研究員、泰地真弘人チームリーダーらの研究チーム※は、新型コロナウイルス感染症(COVID-19)の原因ウイルスである「SARS-CoV-2」のメインプロテアーゼ(Mpro)[1]タンパク質と7種類のヒト免疫不全ウイルス(HIV)プロテアーゼ阻害薬[2]が結合する過程の分子動力学(MD)[3]シミュレーションを行いました。
本研究成果は、新型コロナウイルスの増殖に必須であるMproを標的とする治療薬の開発に役立つと期待できます。
ウイルスは、感染した細胞にウイルスタンパク質を作らせることで増殖します。SARS-CoV-2のMproは、細胞に作らせたタンパク質を適切な箇所で切断して完成させるハサミ(プロテアーゼ)として機能します。MproはHIVのプロテアーゼと類似していることから、既存のHIVプロテアーゼ阻害薬をSARS-CoV-2の治療に応用することが期待されています。
今回、研究チームは、7種類のHIVプロテアーゼ阻害薬について、それぞれMpro表面に接触する過程のMDシミュレーションを行い、結合しやすい場所(結合ポケット)の分類と、プロテアーゼ活性部位への結合しやすさを調べました。さらに、結合ポケットの構造変化の解析によって、この構造が非常に揺らいでいることや、阻害薬と結合した状態でもさまざまな形や配置を取り得ることが分かりました。阻害薬と標的タンパク質の動的な結合を例示した本研究成果は、薬候補分子の発見の新たな可能性を拓くものです。
本研究の生データ[4]はリポジトリ[4] zenodo にて5月13日に公開され、成果は、科学雑誌『Scientific reports』(10月12日号)に掲載されました。

HIV阻害薬(ネルフィナビル=白丸で囲んだ分子)がMproに結合する様子
背景
新型コロナウイルス感染症(COVID-19)は、2019年に中国・武漢で初めて報告されてから現在に至るまで全世界に広がり、世界保健機関(WHO)はパンデミック(世界的大流行)を表明しました。この新型ウイルスによる社会的影響は世界規模に達しており、COVID-19への有効な薬の開発は極めて緊急性の高い、国際的事象となっています。
コロナウイルスは一本鎖RNAをゲノムとして持ち、宿主細胞に感染するとRNAゲノムから長いタンパク質(ポリタンパク質[5])が翻訳されます。このポリタンパク質が切断されることで、それぞれの断片がウイルスの増殖に必要な構造タンパク質や酵素として働きます。COVID-19の原因ウイルス「SARS-CoV-2」が同定されると、複数の機関がウイルスタンパク質の立体構造解析に取り組み、その成果が公的データベースに次々と登録されています注1)。
構造解析に基づく抗ウイルス薬の開発に際して、標的となるタンパク質の一つが、ポリタンパク質の切断を主に触媒するメインプロテアーゼです。メインプロテアーゼの切断活性を阻害すれば、細胞に感染したウイルスの増殖を抑制できると考えられ、メインプロテアーゼに結合する阻害分子の探索が進められています。実際に、ヒト免疫不全ウイルス(HIV)においては、そのプロテアーゼを標的とした薬剤が複数開発され、有効性が示されています。
近年の医薬品開発においては、標的タンパク質に対する薬候補分子の探索・評価にスーパーコンピュータ(スパコン)を活用するインシリコ創薬[6]が注目されています。分子動力学(MD)シミュレーションは、細胞内に近い状況での分子間相互作用を解析する手法であり、水溶液中での分子の動き[7]を考慮することによって高精度な薬物探索が可能となります。
研究チームは、創薬専用スパコン「MDGRAPE-4A[8]」を開発し、2019年11月より実験運用を開始しました注2)。MDGRAPE-4Aは、通常の計算機よりも短時間でタンパク質の構造変化をシミュレートすることを得意とするため、標的タンパク質、薬剤候補分子、水分子の動的構造をサンプリング後、その構造データを使用して高精度な結合親和性予測を行い、現実的な計算時間で高精度なインシリコスクリーニングを実現すると期待されています。これまで、研究チームでは、創薬専用スパコンの速やかに計算できる能力を生かし、COVID-19対策への一環としてMproタンパク質の構造動態をシミュレーションした生データの公開を行っています注3,4)。
注1)最新情報は、大阪大学蛋白質研究所の日本蛋白質構造データバンク(Protein Data BankJapan: PDBj) 新型コロナウイルスの構造情報のページを参照。
注2)2019年11月18日プレスリリース「創薬専用スパコンの開発」
注3)COVID-19 related trajectory data of 10 microseconds all atom molecular dynamics simulation of SARS-CoV-2 dimeric main protease
注4)2020年3月23日プレスリリース「新型コロナウイルス(SARS-CoV-2)メインプロテアーゼの分子動力学シミュレーションデータを公開」
研究手法と成果
研究チームは、新型コロナウイルスのメインプロテアーゼ(Mpro)を阻害する薬分子の開発に役立てるために、HIV阻害薬として既に利用されている7種類の薬分子とMproとの結合過程を分子動力学(MD)計算によりシミュレートしました。MD計算の実行には、理研情報基盤センターHOKUSAIシステムおよび専用計算機MDGRAPE-4Aを使用しました。
シミュレーションに用いたSARS-CoV-2 Mproの立体構造はLiuらの報告注5)をもとにし、溶媒としての水分子数は3万弱、系の総原子数は10万弱としました。7種類の薬分子それぞれについて、Mproの入った水溶液中に薬分子1分子を加えた系の0.2マイクロ秒(1マイクロ秒は100万分の1秒)間のシミュレーションを28試行ずつ行い、Mpro表面に薬分子が取り付く様子を観察しました。全試行(7×28試行)のデータで得られた薬分子とMproの接触データから、Mpro表面の薬が接着しやすい場所(結合ポケット)を分類し、そのうちの一つがMproのハサミとしての機能に直接関わる部位(プロテアーゼ活性部位)に位置することを同定しました(図1)。また、0.2マイクロ秒間にわたる結合の過程を調べたところ、実際に薬分子が活性部位に結合していく様子が確認されました(図2)。
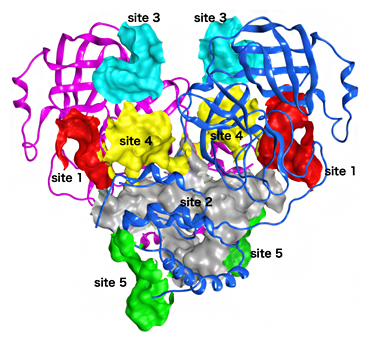
図1 SARS-CoV-2メインプロテアーゼ(Mpro)の結合ポケット
Mproは、二つのサブユニットからなるホモ2量体として機能する。7種類の薬分子の結合データからMpro表面の結合ポケットを分類し、主要な五つのサイト(site1-site5)を示した。赤で示したsite1がプロテアーゼ活性部位と重なる。
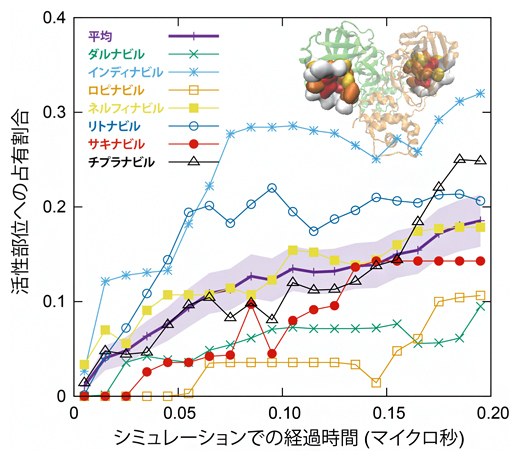
図2 薬分子の活性部位への結合過程
7種類の薬分子を用いたMDシミュレーションにおいて、各薬分子がプロテアーゼ活性部位を占有していた割合を時間経過で見た図。0.2マイクロ秒間の間に、活性部位への結合が増えていることが読み取れる。サンプル数の制約のため、各薬分子の優劣を正確に予測することは難しいが、インディナビル(水色)は有意にダルナビル(緑)よりも親和性が高い傾向を見せている。
次に、薬分子が結合したMproの構造変化を捉えるため、前述のシミュレーションにおいて、各薬分子が最終的に活性部位に結合していた23試行についてMD計算を延長し、1マイクロ秒までの動態を観察しました。その結果、結合ポケットの形状は大きく揺らいでいることが分かりました。さらに3試行について計算時間を6マイクロ秒まで延長したところ、時折、薬分子が結合ポケットの中で向きを変える様子を観察できました(図3)。これは、薬分子と標的タンパク質との結合が、従来考えられてきたよりもはるかに柔軟なものであることを示すものです。
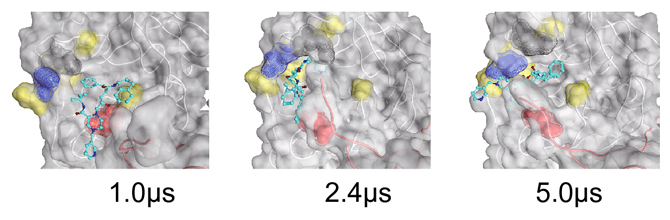
図3 結合ポケットの中で向きを変える薬分子
インディナビル(水色の主鎖で示したスティックモデル)と結合ポケットsite-1(活性部位)の結合を6マイクロ秒間にわたってMDシミュレーションした結果。インディナビルの向きが1.0マイクロ秒(μs)、2.4マイクロ秒、5.0マイクロ秒で大きく変わっている。Mproは空間充填モデルで表し、インディナビルとの結合に関わるアミノ酸残基を赤(166番グルタミン酸)、青(189番グルタミン)、黄(44番システイン、143番グリシン、187番アスパラギン酸、188番アルギニン、190番スレオニン)、灰(49番メチオニン)で示した。これらの位置関係も揺らいでいることが分かる。
今後の期待
現在まで、SARS-CoV-2 Mproに結合するいくつかの抗ウイルス薬候補が見つけられ、その複合体のX線結晶構造解析[9]も行われています。実際に新薬ができるまでには、さらに多くの研究プロセスが必要ですが、本研究のMDシミュレーションを進めて、国内外の創薬研究の進展に寄与したいと考えています。
MDGRAPE-4Aは1日に約1.1マイクロ秒の計算性能を持ち、長時間のMDシミュレーション計算に威力を発揮します。本研究で用いられた計算の生データは誌上発表に先立ち、zenodoレポジトリにて5月13日に公開し、世界の創薬研究者に自由に利用できるようになっています注6)。本研究では、Mproタンパク質の結合ポケットの分類と同時にその形状の柔らかい動態も報告しており、今後のMproをターゲットとした薬分子開発の基礎的データになると考えられます。
今後、研究チームは、薬分子候補の探査および新しい薬分子の開発に向けたデータを取得する目的で、SARS-CoV-2 MproやSARS-CoV-2中にコードされる他の潜在的創薬ターゲットタンパクのシミュレーションをより幅広い薬分子について行っていく予定です。
注6)Molecular dynamics trajectories for SARS-CoV-2 Mpro with 7 HIV inhibitors![]()
補足説明
1.メインプロテアーゼ (Mpro)
プロテアーゼは、タンパク質を加水分解する酵素の総称。コロナウイルスは二つのプロテアーゼを持ち、ポリタンパク質の切断のほとんどを担うものをメインプロテアーゼと呼ぶ。メインプロテアーゼは、二つの同じサブユニットで構成される2量体である。
2.ヒト免疫不全ウイルス(HIV)プロテアーゼ阻害薬
ヒト免疫不全ウィルス(HIV)の治療薬として、HIVの持つリバーストランスクリプターゼ、プロテアーゼ、インテグラーゼに対する活性阻害剤が用いられている。本研究では、プロテアーゼ阻害剤であるダルナビル、インディナビル、ロピナビル、ネルフィナビル、リトナビル、サキナビル、チプラナビルを用いた。HIVはHuman Immunodeficiency Virusの略。
3.分子動力学(MD)
原子間に働く力を計算し、運動方程式を繰り返し解くことで、分子の動きを追跡する方法。分子動力学法の基礎の開発について、2013年のノーベル化学賞が授与されている。MDはMolecular Dynamicsの略。
4.生データ、リポジトリ
今回公開した生データは、溶液中の溶質(メインプロテアーゼ)と溶媒(水分子)および薬分子(HIV阻害薬)を構成する原子1個1個の座標を2.5フェムト秒(2.5×10-15秒)ごとに計算したデータであり、総計85.6マイクロ秒間(8.56×10-5秒)にわたる構造動態データである。
リポジトリは、論文などのデータを共有するために整備されたサーバーを指し、大学などの研究機関が設置したものは、機関リポジトリと呼ばれる。多くのリポジトリでは、インターネットを通じて掲載論文を自由に閲覧できるようになっている。近年は、査読前の論文をリポジトリに投稿してオープンな議論を進める場合も多く、この目的のサーバーはプレプリントサーバーと呼ばれる。今回利用したzenodo![]() は、科学研究データを公開し共有することを推進する目的でECが設立したリポジトリである。
は、科学研究データを公開し共有することを推進する目的でECが設立したリポジトリである。
5.ポリタンパク質
コロナウイルスのRNAゲノムから翻訳される巨大なタンパク質をポリタンパク質と呼ぶ。ポリタンパク質は自らのプロテアーゼ活性により切断され、複数の断片が機能を持つタンパク質となる。
6.インシリコ創薬
細胞生物学的、生化学的な手法を主とする創薬候補物質の探索に対して、コンピュータ(シリコンチップ)の中で行う創薬をインシリコ(in silico)創薬と呼ぶ。
7.水溶液中での分子の動き
生体内のタンパク質の多くは、水に溶けた状態で機能する。すなわち、タンパク質の周りには大多数の水分子が存在し、タンパク質の立体構造や基質との結合に影響を与える。
8.MDGRAPE-4A
1990年より開発が進められている天文学分野での重力(GRAvity)多体問題の計算に特化した専用計算機GRAPE(GRAvity PipE、重力パイプライン)の、分子動力学(Molecular Dynamics: MD)バージョン。MDGRAPE-4Aはその5作目に当たる。開発には、本研究チーム※の他、チョウ コウ(Zhang Hao)研究員、大村一太研究員、小山洋連携促進コーディネーター、西田圭吾大学院生リサーチ・アソシエイトが参加した。
また、日本医療研究開発機構(AMED)創薬支援推進事業-創薬支援インフォマティクスシステム構築「分子シミュレーションによる薬物代謝酵素シトクロム P450(CYP)に対する薬物代謝の予測法開発(研究担当者:泰地真弘人)」、日本学術振興会(JSPS)最先端研究基盤事業「生命動態システム科学研究の推進」、科学研究費補助金基盤研究(A)「次世代分子動力学シミュレーション専用計算機の基盤開発(研究代表者:泰地真弘人)」から一部支援を受け、大正製薬株式会社との共同研究として開発を行った。
9.X線結晶構造解析
構造生物学の手法の一つ。タンパク質の結晶を作製し、その結晶にX線を照射して得られる回折データを解析することにより、タンパク質の内部の原子の立体的な配置を調べる方法。この方法によって、タンパク質の形(立体構造)や内部構造を知ることができる。
研究チーム
理化学研究所 生命機能科学研究センター 計算分子設計研究チーム
研究員 小松 輝久(こまつ てるひさ)
上級研究員 沖本 憲明(おきもと のりあき)
研究員 小山 洋平(こやま ようへい)
研究員 平野 秀典(ひらの よしのり)
技師 森本 元太郎(もりもと げんたろう)
上級技師 大野 洋介(おおの ようすけ)
チームリーダー 泰地 真弘人(たいじ まこと)
原論文情報
KOMATSU, Teruhisa S.; OKIMOTO, Noriaki; KOYAMA, Yohei M.; HIRANO, Yoshinori; MORIMOTO, Gentaro; OHNO, Yousuke; TAIJI, Makoto, “Drug binding dynamics of the dimeric SARS-CoV-2 main protease, determined by molecular dynamics simulation”, Scientific Reports, 10.1038/s41598-020-74099-5
発表者
理化学研究所
生命機能科学研究センター 計算分子設計研究チーム
研究員 小松 輝久(こまつ てるひさ)
上級研究員 沖本 憲明(おきもと のりあき)
チームリーダー 泰地 真弘人(たいじ まこと)
報道担当
理化学研究所 広報室 報道担当

