大腸炎など炎症性腸疾患(IBD)の予防・治療に期待
2022-01-12 生理学研究所
ポイント
①P2Y6Rが難病指定されているIBDの発症に寄与することを明らかにしました。
②緑黄色野菜に多く含まれるスルフォラファンやイベリンが細胞膜表面にあるP2Y6Rと結合し、細胞内への取り込みと分解を促進することで、炎症を抑制することを明らかにしました。
③②はP2Y6R以外の味覚・嗅覚を司る受容体にも共通する機構であることから、今回の発見は、炎症の予防・治療だけでなく、味覚・嗅覚異常のメカニズム解明にもつながる可能性が期待されます。
クローン病や潰瘍性大腸炎に代表される炎症性腸疾患(IBD)は寛解と再燃を繰り返す腸管の慢性炎症を特徴とする原因不明の難治性の疾患です。最新の統計では、クローン病患者数は約7万人、潰瘍性大腸炎患者数は約22万人と推定されており、日本でも増加傾向が続いています。
九州大学大学院薬学研究院の西田基宏教授(生理学研究所・生命創成探究センター教授兼務)と西山和宏講師は、生理学研究所(生命創成探究センター)、東北大学、筑波大学、大阪府立大学、東京工業大学、東京大学との共同研究により、ブロッコリースプラウトなどの緑黄色野菜に多く含まれるスルフォラファンやイベリンが細胞膜表面にある炎症誘導性のGタンパク質共役型受容体「P2Y6R」と結合し、細胞内への取り込みと分解を促進することで、炎症を抑制することを明らかにしました。つまり、P2Y6RがIBDの病態形成の増悪因子であることが分かりました。
細胞の膜表面に存在する受容体タンパク質は、細胞外の様々な物質を感知し、細胞内に情報を伝達する重要な役割を担っています。細胞外の核酸を感知するP2Y6Rは細胞の遊走や貪食を促進する生理機能を有しますが、その作用の増強により炎症が増悪することも知られています。我々は、スルフォラファンがP2Y6Rタンパク質のシステインと直接結合することで、P2Y6Rを細胞膜から解離し、分解を促すことで抗炎症作用を発揮することを見出しました。P2Y6Rの細胞内取り込みは、既存の制御機構とは異なるシステイン酸化に依存した機構であり、P2Y6R以外の味覚・嗅覚を司る受容体にも共通する機構であることも明らかにしました。今回の発見は、炎症の予防・治療だけでなく、味覚・嗅覚異常のメカニズム解明にもつながる可能性が期待されます。
本研究結果は、米国医学誌が発行する誌「Science Signaling」にオンライン版にて掲載される予定です。
研究者からひとこと:
薬学研究院では、病気の原因となるタンパク質と直接化学的に結合し、その機能を選択的に阻害する「コバレント創薬」にも取り組んでいます。今回の発見は、心血管病や炎症性腸疾患などの予防・治療薬の開発につながるだけでなく、農学や歯学、環境医学などとの異分野連携・融合にも発展する可能性が期待されます。

(参考図)
青色:2012年ノーベル化学賞受賞研究としても有名な既存の受容体タンパク質取り込み(負のフィードバック)経路。赤色:本研究で見出したシステイン硫黄修飾を介した新奇受容体取り込み&分解経路。動物個体において、本機構が機能性食品成分による抗炎症効果などに重要な役割を果たすことも明らかになった。
<研究の内容>
クローン病や潰瘍性大腸炎に代表される炎症性腸疾患(IBD)は寛解と再燃を繰り返す腸管の慢性炎症を特徴とする原因不明の難治性の疾患です。IBDは腸内細菌や食生活などの環境要因と、遺伝要因、そして体内の免疫異常が重なって発症する疾患と考えられています。
Gタンパク質共役型受容体(GPCR)は様々な生理機能や疾患形成に関わる膜タンパク質です。私たちのグループはこれまで、GPCRの1種であるプリン作動性受容体P2Y6Rが加齢依存的に発現増加し、高血圧や心不全の発症に寄与することを報告しました。一方で、IBD患者でもP2Y6Rの発現上昇が報告されていましたが、IBDの病態に対する役割は不明でした。
今回私たちは、IBDモデルであるデキストラン硫酸ナトリウム(DSS)誘発性大腸炎とP2Y6R欠損マウスを用いた検討より、P2Y6R欠損マウスでは野生型マウスと比較して、大腸炎の進行が抑制されていたことを明らかにしました。つまり、P2Y6RがIBDの病態形成の増悪因子であることが明らかになりました(図1)。さらに、P2Y6Rを阻害する化合物を探索したところ、機能性食品(ブロッコリースプラウト)に含まれるスルフォラファンなどのイソチオシアネート化合物(ITCs)が、P2Y6Rの内在化およびプロテアソーム系分解を介して阻害することを見出しました(図2)。GPCRの細胞内取り込み機構に関しては、2012年のノーベル科学賞受賞者であるLerkowitz教授らがβアレスチンを介する経路を発見しましたが、いくつかのGPCRはβアレスチン感受性が低いこともわかっていました。今回、我々は、βアレスチン低感受性のP2Y6Rを用いて、ITCがP2Y6Rの細胞内第3ループにあるシステイン(Cys220)のSH基と結合することで、第2ループのリジン(Lys137)のユビキチン化が誘導されることを明らかにしました。P2Y6RのCys220をセリンに置換した変異体マウス(C220S)では野生型マウスと比較して、逆に大腸炎の進行が促進されました。さらに、Cys修飾によるLysユビキチン化を介した内在化・分解機構(redox-dependent alternative internalization:REDAI)は、P2Y6R以外のGPCRでも保存されていることが確認されました(参考図)。
<今後の展開と治療応用への期待>
今回の結果より、過剰なP2Y6Rの発現によるシグナルの増加がIBD病態を進行させる可能性が示されました。さらに、機能性食品に含まれるスルフォラファンなどがREDAIシステムを介してP2Y6Rを分解し、炎症を抑制することも明らかとなりました。本研究の成果により、これまでのGPCRの制御機構とは全く異なった新奇内在化機構の存在が明らかとなりました。GPCR は大腸炎をはじめ心不全や高血圧など様々な疾患制御に関連するため、REDAIシステムを利用した治療法・医薬品開発が健康長寿社会の実現に大きく貢献するものと期待されます。
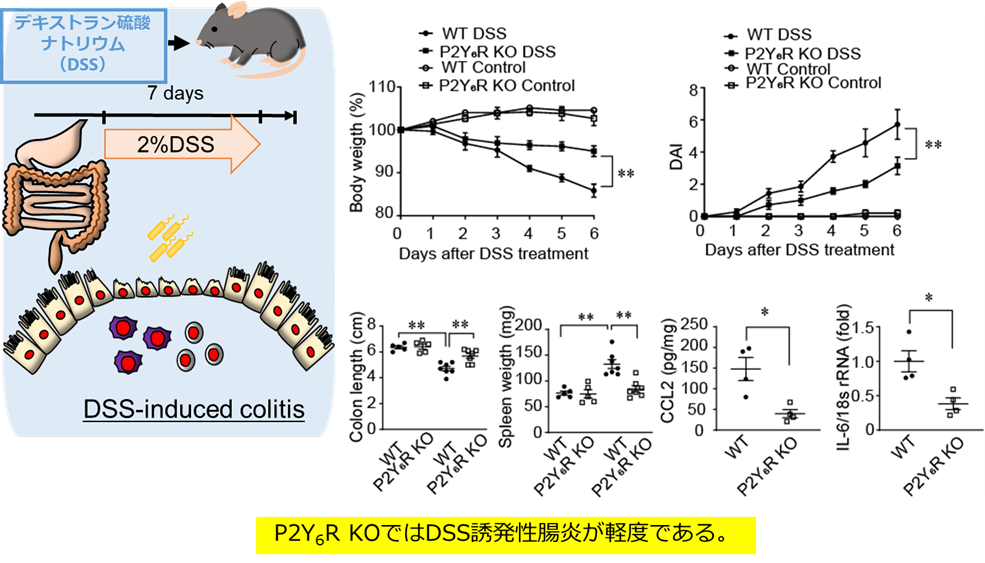 図1 P2Y6Rは腸炎の発症に関連する。
図1 P2Y6Rは腸炎の発症に関連する。
P2Y6R欠損マウスでは野生型マウスと比較して、大腸炎の進行が抑制されていたことからP2Y6RがIBDの発症に寄与することを明らかにしました。
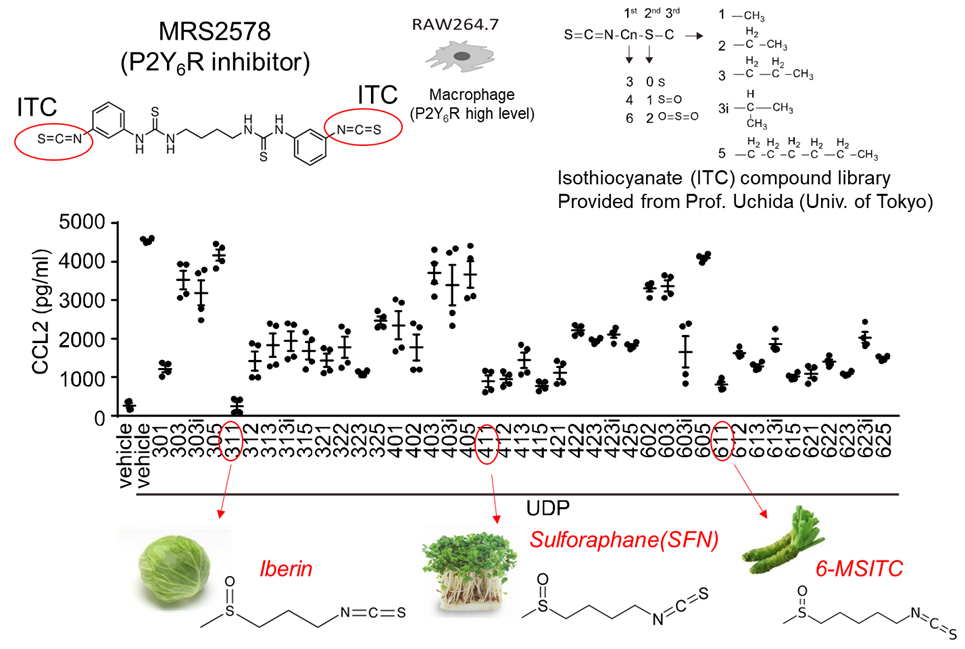 図2 ブロッコリースプラウトに含まれるスルフォラファンがP2Y6Rを抑制する。
図2 ブロッコリースプラウトに含まれるスルフォラファンがP2Y6Rを抑制する。
P2Y6Rを阻害する化合物を探索したところ、機能性食品に含まれるスルフォラファンなどのイソチオシアネート化合物(ITCs)が、P2Y6Rの内在化およびプロテアソーム系分解を介して阻害することを見出しました。
【主な共同研究者と所属機関】
・西田 基宏 教授
九州大学大学院薬学研究院 生理学分野
自然科学研究機構 生命創成探究センター/生理学研究所(兼務)
・西山 和宏 講師、加藤 百合 助教、
九州大学大学院薬学研究院 生理学分野
・西村 明幸 特任准教授、田中 智弘 特任助教
自然科学研究機構生命創成探究センター/生理学研究所
・柴田 貴広 教授、
名古屋大学大学院生命農学研究科
・田中 浩士 准教授
東京工業大学大学院物質理工学院応用化学系
・東 泰孝 教授、
大阪府立大学大学院生命環境科学研究科
・居原 秀 教授、
大阪府立大学大学院理学系研究科
・熊谷 嘉人 教授
筑波大学大学院医学医療系環境生物学分野
・赤池 孝章 教授、
東北大学大学院医学系研究科
・Philip Eaton教授、
Queen Mary University of London
・内田 浩二 教授、
東京大学大学院農学生命科学研究科
【謝辞】
本研究は国立研究開発法人科学技術振興機構(JST)・戦略的創造研究推進事業(CREST)の研究領域「多細胞間での時空間的相互作用の理解を目指した定量的解析基盤の創出」における研究開発課題「超硫黄フラックス解析基盤の創出による筋頑健性構築」(代表:西田 基宏教授)の一環で行われたと共に、日本学術振興会の科学研究費補助金、文部科学省の新学術領域「酸素生物学」、喫煙科学研究財団、小野医学研究財団などの研究助成、ならびに日本医療研究開発機構(AMED)「創薬等ライフサイエンス研究支援基盤事業(九大拠点)」による支援を受けて行われました。
【論文情報】
タイトル:Redox-dependent internalization of purinergic P2Y6 receptor limits colitis progression.
掲載誌:Science Signaling. 2022
著者名:Kazuhiro Nishiyama, Akiyuki Nishimura, Kakeru Shimoda, Tomohiro Tanaka, Yuri Kato, Takahiro Shibata, Hiroshi Tanaka, Hitoshi Kurose, Yasu-Taka Azuma, Hideshi Ihara, Yoshito Kumagai, Takaaki Akaike, Philip Eaton, Koji Uchida and *Motohiro Nishida.
DOI: 10.1126/scisignal.abj0644
【お問合せ先】
<研究に関するお問合せ先>
九州大学大学院薬学研究院 生理学分野 教授 西田 基宏
九州大学大学院薬学研究院 生理学分野 講師 西山 和宏
<報道に関するお問合せ先>
九州大学広報室
自然科学研究機構 生理学研究所 研究力強化戦略室
自然科学研究機構 生命創成研究センター 研究連携推進室

